
Komar Anopheles stephensi krótko po pobraniu krwi od człowieka (kropla krwi jest wydalana jako nadmiar). Komar ten jest wektorem malarii, a zwalczanie komarów jest skutecznym sposobem na ograniczenie jej występowania. Domena publiczna.
Streszczenie
Ludzki pasożyt Plasmodium malariae ma krewnych zakażających afrykańskie małpy człekokształtne ( Plasmodium rodhaini ) i małpy Nowego Świata ( Plasmodium brasilianum ), ale jego pochodzenie pozostaje nieznane. Stosując nowatorskie podejście do charakteryzowania sekwencji powiązanych z P. malariae u dzikich i żyjących w niewoli afrykańskich małp człekokształtnych, odkryliśmy, że grupa ta składa się z trzech odrębnych linii, z których jedna reprezentuje wcześniej nieznany, wysoce zróżnicowany gatunek zakażający szympansy, bonobo i goryle w całej Afryce Środkowej. Druga linia pochodząca od małp jest znacznie bliżej spokrewniona z trzecią, zakażającą człowieka linią P. malariae , ale wykazuje niewiele dowodów na wymianę genetyczną z nią i dlatego prawdopodobnie reprezentuje odrębny gatunek. Ponadto poziomy i charakter polimorfizmów genetycznych w P. malariae wskazują, że powstał on w wyniku odzwierzęcej transmisji afrykańskiego pasożyta małp człekokształtnych, przypominając pochodzenie P. falciparum . Natomiast P. brasilianum mieści się w zakresie promieniowania ludzkiej P. malariae i w związku z tym odzwierciedla niedawną antropopresję.
Podobna treść jest oglądana przez innych

Wstęp
Plasmodium malariae jest jednym z sześciu gatunków Plasmodium , które powszechnie powodują malarię u ludzi1. Chociaż geograficznie szeroko rozpowszechniony w tropikalnych i subtropikalnych regionach endemicznych malarii2,3, P. malariae jest najsłabiej scharakteryzowanym pasożytem ludzkim, częściowo dlatego, że osiąga jedynie bardzo niskie poziomy parazytemii, jest ogólnie kojarzony z łagodną postacią choroby lub jej brakiem4 i często współwystępuje z innymi pasożytami malarii w zakażeniach wielogatunkowych 5. W przeciwieństwie do innych pasożytów ludzkich, P. malariae infekuje tylko dojrzałe czerwone krwinki z cyklem wewnątrzerytrocytowym trwającym 72 godziny, co powoduje charakterystyczną gorączkę kwartan 4. Jednak pasożyt może również przetrwać przewlekle i nawracać lata lub dekady po pierwotnym zakażeniu 2 , 3. Z powodu braku systematycznych badań spowodowanych postrzeganym niskim obciążeniem chorobą, częstość występowania P. malariae jest prawdopodobnie niedoszacowana 6 , 7 , 8 .
Podczas gdy pochodzenie głównych ludzkich pasożytów malarii P. falciparum i P. vivax zostało niedawno prześledzone do pasożytów infekujących afrykańskie małpy człekokształtne 9 , 10 , 11 , 12 , 13 , 14 , 15, ewolucyjna historia P. malariae pozostaje nieznana, pomimo od dawna znanego istnienia bliskich krewnych u naczelnych innych niż człowiek 16 . Na początku XX wieku zaobserwowano pasożyta kwartanowego, morfologicznie nieodróżnialnego od P. malariae, infekującego dzikie szympansy, któremu nadano nazwę P. rodhaini 17 . Jednak w późniejszych badaniach transmisji P. rodhaini został pomyślnie przeniesiony z szympansów na ludzi poprzez szczepienie krwi 18 , podczas gdy P. malariae został przeniesiony z ludzi na szympansy przez krew 19 i zakaźne komary 20 . Ten pozorny brak specyficzności żywiciela sugerował, że pasożyty małp człekokształtnych i ludzi były członkami tego samego gatunku16, ale nie dało się tego zweryfikować, ponieważ szczepy P. rodhaini nie były hodowane i w związku z tym nigdy nie zostały scharakteryzowane genetycznie.
Niedawno badania genetyczne zostały zastosowane do krewnych P. malariae zakażających małpy człekokształtne. Po pierwsze, analizy sekwencji mitochondrialnego DNA (mtDNA) pochodzących z próbek kału dziko żyjących szympansów, goryli i bonobo wskazały na istnienie dwóch odrębnych linii związanych z P. malariae zakażających małpy człekokształtne: jednej, która jest blisko spokrewniona z pasożytem ludzkim i drugiej, która jest znacznie bardziej rozbieżna9 , 21 , 22. Potwierdzając to, sekwencje mtDNA uzyskane z próbek komarów ( Anopheles vinckei ) , które żywią się małpami człekokształtnymi, również wydawały się należeć do dwóch rozbieżnych linii23 . Niedawno sekwencje genomu, jedną prawie kompletną (PmlGA01) i jedną częściową (PmlGA02), odzyskano z krwi dwóch szympansów żyjących w rezerwacie przyrody w Gabonie24. Chociaż analizy całego genomu wykazały bliskie pokrewieństwo z P. malariae, te pasożyty „ podobne do P. malariae ” uznano za odrębny gatunek, który mógł oddzielić się od P. malariae ponad 3 miliony lat temu24 . Jednakże PmlGA01 i PmlGA02 różniły się od siebie tak samo, jak od P. malariae , a badanie nie dostarczyło żadnych informacji na temat pokrewieństwa tych dwóch pasożytów z dwoma rozbieżnymi liniami pasożytów małp człekokształtnych ujawnionymi przez mtDNA.
Wreszcie, inna linia pokrewna P. malariae, P. brasilianum, zaraża małpy Nowego Świata w całej Ameryce Środkowej i Południowej. Charakterystyka molekularna P. brasilianum, aczkolwiek tylko w ograniczonej liczbie loci genetycznych, wykazała , że jest ona niemal identyczna z P. malariae , co ponownie sugeruje, że te dwie linie mogą w rzeczywistości być jednym gatunkiem4 , 24 , 25 , 26. Ponadto, P. brasilianum był przenoszony z małp Nowego Świata na ludzi przez komary i przenoszony z powrotem na małpy poprzez zakażoną krew16 , a małpy Nowego Świata mogły zostać eksperymentalnie zakażone P. malariae od człowieka27 . Odkrycia te sugerują, że pasożyt małp Nowego Świata powstał jako antroponoza od ludzi zakażonych P. malariae28 . Jednakże porównanie różnorodności genetycznej doprowadziło niektórych autorów do wniosku, że możliwy jest również scenariusz przeciwny, tzn. że P. malariae pochodzi od P. brasilianum 29 , 30 , 31.
Aby określić liczbę gatunków spokrewnionych z P. malariae i zakres ich żywicieli, zastosowaliśmy PCR z rozcieńczeniami granicznymi (Low Dilution PCR) do wygenerowania nowych sekwencji z próbek kału i krwi dziko żyjących i żyjących w niewoli szympansów i goryli. Opracowaliśmy również nowe podejście do identyfikacji sekwencji spokrewnionych z P. malariae w opublikowanych bazach danych genomu małp człekokształtnych z mieszanymi zakażeniami Plasmodium . W niniejszej pracy pokazujemy, że klad pasożytów spokrewnionych z P. malariae obejmuje trzy odrębne linie rozwojowe, z których jedna reprezentuje nieznany wcześniej, odległy gatunek, który zakaża dziko żyjące szympansy, bonobo i goryle w całej Afryce Środkowej i Zachodniej. Wykazujemy również, że pasożyty „ podobne do P. malariae ” 24 reprezentują drugi gatunek zarażający małpy człekokształtne, który jest znacznie bliżej spokrewniony z linią zakażającą człowieka, ale brakuje dowodów na wymianę genetyczną z pasożytem ludzkim. Co więcej, porównanie polimorfizmów genetycznych obu linii wskazuje, że pasożyt ludzki przeszedł poważne wąskie gardło genetyczne, po którym nastąpiła gwałtowna ekspansja populacji. Wreszcie, wykazujemy, że P. brasilianum nie reprezentuje odrębnej linii, ale mieści się w zakresie promieniowania szczepów P. malariae u ludzi. Wyniki te wskazują zatem na pochodzenie kladu spokrewnionego z P. malariae w Afryce i wskazują, że pasożyt ludzki powstał w wyniku zmiany żywiciela pasożyta małpy człekokształtnej, podczas gdy P. brasilianum powstał po przeniesieniu pasożyta ludzkiego na małpy Nowego Świata.
Wyniki
Trzy odrębne linie rozwojowe w grupie spokrewnionej z P. malariae
Chociaż od dawna wiadomo, że dzikie małpy są nosicielami pasożytów spokrewnionych z P. malariae17 ( zdefiniowanych jako grupa pasożytów, które mają znacznie większe podobieństwo sekwencji do P. malariae niż do jakiegokolwiek innego gatunku Plasmodium , a do których należy P. brasilianum i sam pasożyt ludzki, patrz Notatka uzupełniająca 1 ), scharakteryzowano tylko kilka zakażeń. Poprzednie badania terenowe dzikich małp i ich komarów wykazały obecność pasożytów spokrewnionych z P. malariae9 , 21 , 22 , 23 , 32 , ale odzyskano bardzo ograniczoną liczbę sekwencji, pochodzących niemal w całości z pojedynczego genu mitochondrialnego ( cytB ). Aby zwiększyć liczbę sekwencji pasożytów do badań filogenetycznych, wybraliśmy próbki krwi i kału małp , w których wcześniej diagnostyczna reakcja PCR wykazała obecność P. malariae , i poddaliśmy je amplifikacji pojedynczego genomu (SGA) ukierunkowanej na różne regiony genomowe. Metoda SGA amplifikuje pojedyncze matryce, unikając w ten sposób generowania sekwencji pasożytów z artefaktami polimerazy Taq , w tym chimer wytworzonych w wyniku rekombinacji, co jest istotne w przypadku próbek z infekcjami wielogatunkowymi. Dzięki temu podejściu udało nam się zamplifikować dodatkowe sekwencje mtDNA i genów jądrowych ( ldh i asl ). Chociaż większość z nich pochodziła z próbek krwi szympansów z sanktuariów, sekwencje związane z P. malariae uzyskano również od jednego szympansa i dwóch goryli pobranych na wolności (tabele uzupełniające 1 i 2 ). Wygenerowaliśmy również nowo zmontowane genomy mtDNA dla dwóch pasożytów szympansów „podobnych do P. malariae ” – GA01 i GA02.
Analizy filogenetyczne zarówno nowo uzyskanych ( N = 21), jak i wcześniej opublikowanych ( N = 44) sekwencji pasożytów wykazały, że P. malariae i pasożyty naczelnych spokrewnione z P. malariae należą do dwóch bardzo rozbieżnych linii w drzewach pochodzących zarówno z sekwencji genów mtDNA (Rys. 1a , Ryc. uzupełniający 1 ), jak i jądrowych (Rys. 1c, d ). Jedna z nich (początkowo określana jako M1) obejmowała wszystkie pasożyty P. malariae u ludzi, wraz z P. brasilianum i niektórymi próbkami od małp człekokształtnych. Druga linia (oznaczana jako M2, patrz Notatka uzupełniająca 1 ) była dość odlegle spokrewniona z P. malariae, chociaż wyraźnie bliższa P. malariae niż jakimkolwiek innym wcześniej opisanym gatunkom Plasmodium (patrz poniżej). Linia M2 obejmowała sekwencje uzyskane z próbek pobranych od goryli, szympansów i bonobo, a także wszystkie 10 sekwencji spokrewnionych z P. malariae, uzyskanych wcześniej od komarów żywiących się małpami człekokształtnymi 23 (ryc. 1a ).
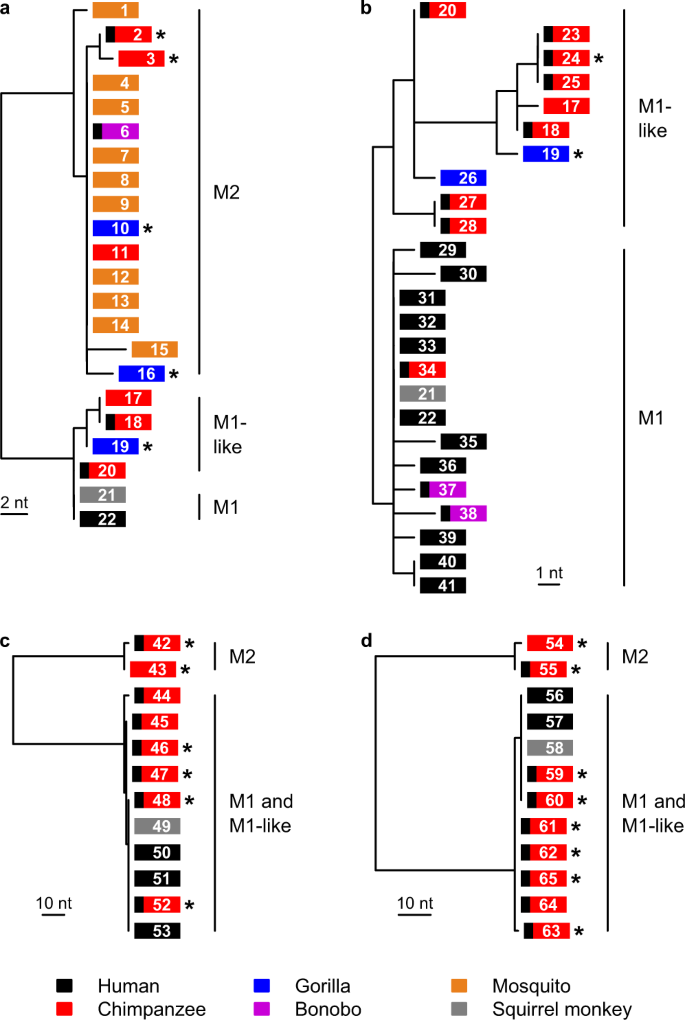
a Drzewo maksymalnego prawdopodobieństwa wyprowadzone z fragmentu mitochondrialnego DNA cytB o długości 641 pz ; drzewo to obejmuje wszystkie dostępne sekwencje M2 cytB , ale dla przejrzystości uwzględnia tylko kilka reprezentatywnych sekwencji M1 i podobnych do M1. b Drzewo maksymalnego prawdopodobieństwa wyprowadzone z fragmentu mitochondrialnego cytB / cox1 o długości 2038 pz ; drzewo to obejmuje wszystkie dostępne sekwencje o wystarczającej długości z tych linii i zostało wygenerowane z dłuższego dopasowania obejmującego region w części a (patrz Dodatkowa Rys. 1 ). c Drzewo maksymalnego prawdopodobieństwa wyprowadzone z fragmentu asl o długości 682 pz . d Drzewo maksymalnego prawdopodobieństwa wyprowadzone z fragmentu ldh o długości 724 pz . Pola reprezentują sekwencje i są pokolorowane zgodnie z żywicielem, od którego pochodzi próbka, jak wskazano w legendzie. Czarny pasek po lewej stronie pola wskazuje próbkę od małpy człekokształtnej żyjącej w niewoli lub w sanktuarium; gwiazdki oznaczają nowo wyprowadzone sekwencje; liczby w polach podają kod sekwencji (Dodatkowa Tabela 2 ). Wsparcie Bootstrap z 1000 replikacji wyniosło 100% dla węzłów oddzielających M2 od M1 i węzłów podobnych do M1 oraz dla węzła oddzielającego M1 od węzłów podobnych do M1 w części b . Podziałka wskazuje liczbę zmian nukleotydów.
Dokładniejsze badanie sekwencji M1 wskazało, że można je podzielić na dwie linie. W drzewie pochodzącym z naszego najdłuższego dostępnego dopasowania mtDNA (~2 kb), dla którego mieliśmy liczne próbki (Rys. 1b , Rys. uzupełniający 1 , Tabele uzupełniające 2 i 3 ), mogliśmy wyróżnić jeden klad obejmujący wszystkie ludzkie sekwencje P. malariae , P. brasilianum i kilka sekwencji od małp człekokształtnych w niewoli, w których sekwencje nie różniły się o więcej niż dwa nukleotydy od konsensusowej sekwencji P. malariae ; zachowaliśmy termin M1 dla tego kladu (patrz Notatka uzupełniająca 1 ). Pozostałe sekwencje inne niż M2 wykazały większą różnorodność i obejmowały próbki pochodzące zarówno od małp dzikich, jak i żyjących w niewoli, w tym dwa szczepy pochodzące od szympansów („ P. malariae -like” GA01 i GA02), dla których wyprowadzono sekwencje genomu24 ; W niniejszym tekście określamy je mianem M1-podobnych, aby uniknąć pomyłki z terminem „ podobne do P. malariae ” w literaturze23 , 24 (patrz uwaga uzupełniająca 1 ). Chociaż szczepy M1 i M1-podobne nie zostały rozdzielone w drzewach genealogicznych uzyskanych z częściowych sekwencji genów jądrowych (ryc. 1c, d ), późniejsze porównania sekwencji genomu jądrowego potwierdziły, że te dwie linie rozwojowe są odrębne .
M2 reprezentuje nowy gatunek spokrewniony z P. malariae , który zaraża małpy człekokształtne w całej Afryce
Aby lepiej scharakteryzować linię M2, najpierw zbadaliśmy jedną próbkę krwi pobraną od szympansa z sanktuarium (SYptt92, sekwencje 2 i 55 na ryc. 1 ), która dostarczyła częściowych sekwencji mitochondrialnych i jądrowych, które skupiły się w obrębie M2. Podjęliśmy próbę selektywnej amplifikacji całego genomu (SWGA 11 , 33 ) DNA z pełnej krwi, ale nie udało nam się uzyskać wystarczającej liczby sekwencji. Jako alternatywne podejście, zapytaliśmy, czy projekty sekwencjonowania ukierunkowane na inne gatunki Plasmodium małp człekokształtnych mogły amplifikować sekwencje pasożytów M2 jako produkt uboczny; uznaliśmy, że jest to możliwe, ponieważ M1 i M2 były wcześniej obserwowane jako koinfekcje z innymi gatunkami Plasmodium 9 , 24 . Przeszukaliśmy zatem bazy danych sekwencji pasożytów z opublikowanych zakażeń Laverania 13 (13 próbek) i P. vivax -like 15 (siedem próbek) szympansów i goryli pod kątem obecności sekwencji związanych z P. malariae . Zmapowaliśmy wszystkie odczyty na połączoną sekwencję referencyjną Plasmodium , w tym genom referencyjny P. malariae PmUG01, a następnie przeprowadziliśmy de novo montaż kontigów par odczytów, które mapowały się na P. malariae i wyrównaliśmy kontigi z montażem genomu podobnego do M1 PmlGA01 24 („ P. malariae -like”). To podejście zidentyfikowało jedną próbkę (PGABG03 13 ) od szympansa w gabońskim rezerwacie dzikiej przyrody, która dostarczyła kontigi z bimodalnym rozkładem tożsamości nukleotydów do odniesienia podobnego do M1 (rys. 2a ). Jeden szczyt obejmował kontigi wykazujące ponad 97% identyczności nukleotydów z PmlGA01, podczas gdy drugi, szerszy szczyt skupiał się wokół 88% identyczności, co sugeruje koinfekcję pasożytami podobnymi do M1 i M2. Wysokiej jakości kontigi z tego drugiego szczytu (szczegóły dotyczące sposobu ich selekcji znajdują się w sekcji Metody) dopasowaliśmy do genomu referencyjnego P. malariae i oznaczonych genów metodą automatycznego transferu, a następnie ręcznej kuracji. Ponieważ przygotowanie bibliotek odczytów z próbki krwi PGABG03 obejmowało etap amplifikacji całego genomu 13 , mogły zostać wygenerowane fragmenty DNA będące mozaikami koinfekujących genomów pasożytów podobnych do M1 i M2. Dlatego przeprowadziliśmy kilka etapów kontroli jakości w celu zidentyfikowania i usunięcia mozaikowych kontigów i innych możliwych błędów (szczegóły w sekcji Metody). Te etapy montażu wygenerowały częściową sekwencję genomu (497 kb) z ortologami 290 P. malariae.geny adnotowane; jednak większość sekwencji genów była niekompletna i było tylko 26 genów pełnej długości. Chociaż żaden z dwóch genów jądrowych amplifikowanych przez SGA nie został w całości pokryty tą częściową sekwencją genomu, zidentyfikowaliśmy 11 odczytów mapujących jeden z nich ( asl ), co dało sekwencję konsensusową z tylko dwoma różnicami od jednego z amplikonów M2 (Rys. 1b , Ryc. uzupełniający 2 ). Chociaż tę sekwencję asl należy traktować ostrożnie ze względu na niskie pokrycie odczytów, to podobieństwo wskazuje, że rozbieżny genom pokrewny P. malariae uzyskany z biblioteki odczytów PGABG03 rzeczywiście należy do linii M2.
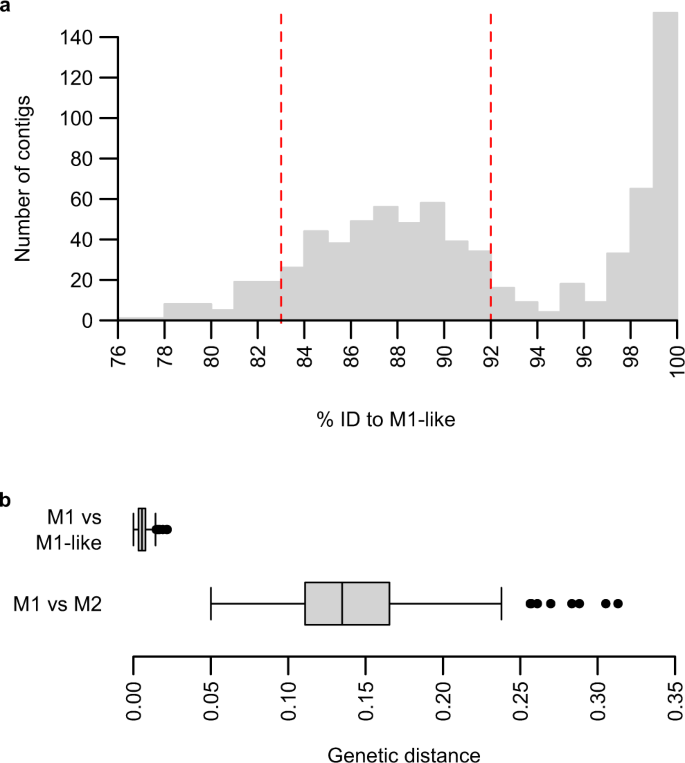
a Tożsamość do montażu genomu podobnego do M1 PmlGA01 kontigów de novo zmontowanych z odczytów sekwencjonowania PGABG03 zmapowanych na P. malariae . Aby poprawić czytelność, jeden kontig o identyczności nukleotydów wynoszącej 70% został pominięty na wykresie. Konigi o identyczności nukleotydów z PmlGA01 wynoszącej od 83% do 92% (przerywane czerwone linie) przyjęto za pochodzące z M2 i wykorzystano do dalszego montażu; kontigi o identyczności od 92% do 97% lub poniżej 83% identyczności zostały ocenione ręcznie i w razie potrzeby uwzględnione jako kontigi M2. b Wykres pudełkowy rozbieżności nukleotydów między M1 i podobnymi do M1 oraz między M1 i M2; n = 285 genów, łącznie 350 kb wyrównanej sekwencji. Pionowa linia środkowa wskazuje medianę; pole wskazuje zakres interkwartylowy; wąsy wskazują 1,5x zakresu interkwartylowego; czarne kółka oznaczają wartości odstające.
Aby scharakteryzować związek między nowymi gatunkami Plasmodium i innymi pasożytami malarii, dopasowaliśmy 285 genów oznaczonych w M2 do ich ortologów M1 i podobnych do M1, używając sekwencji z zestawu PmUG01 i sekwencji uzyskanych przez wywołanie polimorfizmu pojedynczego nukleotydu (SNP) ze szczepu podobnego do M1 GA01 24 . Obliczyliśmy skorygowane dywergencje nukleotydowe dla pojedynczych genów (rys. 2b , rys. uzupełniający 3 ) i dla wszystkich sekwencji łącznie, uzyskując odległość genetyczną między M1 i M2 wynoszącą 13,5%, w porównaniu z 0,6% między M1 i podobnymi do M1. Następnie wygenerowaliśmy dopasowania aminokwasów dla 186 genów, dla których ortologi można było zidentyfikować z gatunków pasożytów ssaków z całego rodzaju Plasmodium , obliczyliśmy dywergencje aminokwasów między parami spokrewnionych pasożytów (tabela 1 ) i skonstruowaliśmy drzewo białkowe (rys. 3 ). Analiza ta potwierdziła, że M2 grupuje się z innymi liniami ewolucyjnymi spokrewnionymi z P. malariae , M1 i M1-like, ale jest z nimi jedynie odlegle spokrewniona. Co więcej, rozbieżność aminokwasów między M1 i M2 (8,5%) była porównywalna z rozbieżnością w obrębie linii P. vivax (9,0% między P. vivax i P. knowlesi ) i większa niż w obrębie innych linii ssaczych Plasmodium (Tabela 1 ). Tak duża rozbieżność wskazuje, że M2 odzwierciedla dawny podział w linii ewolucyjnej spokrewnionej z P. malariae i dlatego powinna być uznana za odrębny gatunek.

Drzewo o maksymalnym prawdopodobieństwie zostało utworzone z dopasowań białek. Sekwencje nukleotydowe dla 186 genów z ortologami jeden do jednego we wszystkich 14 genomach zostały przetłumaczone i dopasowane, a następnie dopasowania genów zostały oczyszczone i połączone w jedno dopasowanie, z którego zbudowano drzewo. Pięć głównych linii jest oznaczonych; nazwy dwóch odpowiadających podrodzajom są napisane kursywą. Drzewo zostało zakorzenione na gałęzi do Laverania 36. Nowa , rozbieżna linia pochodząca od małpy człekokształtnej M2 jest wyświetlana na górze drzewa na czerwono. Wsparcie Bootstrap z 1000 replikacji wyniosło co najmniej 99% dla wszystkich węzłów, z wyjątkiem węzła oddzielającego linie Malariae i Vivax od reszty drzewa. Podziałka reprezentuje 0,05 podstawień/miejsce.
Pasożyty M1 i podobne do M1 reprezentują dwie linie nierekombinujące
Analizy sekwencji mtDNA (ryc. 1b ) sugerowały rozróżnienie między sekwencjami P. malariae pochodzącymi od ludzi (M1) a sekwencjami pokrewnymi P. malariae uzyskanymi od dzikich małp człekokształtnych (podobnymi do M1), chociaż sekwencje pochodzące od małp człekokształtnych żyjących w niewoli należały zarówno do kladów M1, jak i M1-podobnych. To rozróżnienie nie było widoczne w porównaniach sekwencji genów jądrowych (ryc. 1c, d ), które wykazywały mniejszą zmienność niż sekwencje mtDNA. Zrozumienie relacji między tymi dwiema liniami jest kluczowe dla wyjaśnienia potencjału odzwierzęcego pasożytów pochodzących od dzikich małp człekokształtnych, dlatego podjęliśmy próbę zwiększenia próbkowania genomów podobnych do M1. Wykonaliśmy SWGA 11 , 33 na dwóch próbkach pozytywnych pod względem P. malariae , jednej pochodzącej od kameruńskiego szympansa z sanktuarium (MOpte51017) i drugiej od dziko żyjącego szympansa (Ptv_Leo) z Wybrzeża Kości Słoniowej, który zmarł na chorobę zakaźną. Specjalnie wybraliśmy te próbki, ponieważ nie zawierały innych gatunków Plasmodium i dlatego mogły zostać amplifikowane bez ryzyka generowania rekombinantów międzygatunkowych. Każdą próbkę amplifikowano wielokrotnie przy użyciu różnych zestawów starterów SWGA, a poszczególne reakcje SWGA połączono przed sekwencjonowaniem MiSeq. Odczyty z MOpte51017 i Ptv_Leo zostały zmapowane na genom referencyjny P. malariae , obejmujący odpowiednio 23,6 Mb i 0,3 Mb (Tabela uzupełniająca 4 ). Dla porównania, dwa opublikowane genomy podobne do M1 obejmowały 20,4 Mb i 6,2 Mb tego samego odniesienia (Tabela uzupełniająca 4 ).
Sekwencje SGA asl i ldh z MOpte51017 były identyczne z sekwencjami z P. malariae (sekwencja 52 na ryc. 1c i sekwencja 60 na ryc. 1d ), co sugeruje, że ten szympans w niewoli był zakażony pasożytem ludzkim, chociaż na podstawie tych dwóch krótkich sekwencji nie można było wykluczyć szczepu podobnego do M1, szczególnie blisko spokrewnionego z M1. Chociaż próbka Ptv_Leo została pobrana od dzikiego małpy człekokształtnej, wcześniej zsekwencjonowany amplikon cytB był niewystarczający do ustalenia, czy ten pasożyt reprezentował infekcję M1, czy podobną do M122,32 . Aby lepiej scharakteryzować te próbki, połączyliśmy nowo uzyskane sekwencje genomu z danymi pochodzącymi z opublikowanych danych pasożytów M1 pochodzących od człowieka (tj. P. malariae ) 24 , 34 , 35 ( N = 22) oraz pasożytów podobnych do M1 pochodzących od szympansów24 ( N = 2 ), identyfikując SNP poprzez porównanie z genomem referencyjnym P. malariae , który również uwzględniliśmy w analizie. Sekwencje genomu różniły się jakością i zakresem pokrycia genomu referencyjnego P. malariae (Tabela uzupełniająca 4 ), przy czym wszystkie szczepy obejmowały jedynie niewielką część genomu referencyjnego.
Sieć filogenetyczna wygenerowana z SNP w regionach genomu pokrytych przez wszystkie cztery genomy pasożytów pochodzących od szympansów i sześć genomów pasożytów pochodzących od ludzi o najwyższym pokryciu ujawniła monofiletyczną linię M1, która obejmowała MOpte51017 (Rys. 4a ), potwierdzając, że szympans w niewoli został rzeczywiście zakażony pasożytem pochodzenia ludzkiego. Natomiast sekwencja Ptv_Leo była podobna do dwóch wcześniej scharakteryzowanych genomów podobnych do M1 (GA01 i GA02) tym, że była zupełnie odmienna od kladu M1, co jest zgodne z tym, że jest to pasożyt szympansów nabyty na wolności. Te trzy genomy podobne do M1, plus klad M1, były w przybliżeniu równoodległe (Rys. 4a ), co sugeruje, że przodek kladu M1 był członkiem linii podobnej do M1.

a Sieć filogenetyczna oparta na cechach, pokazująca powiązania między wszystkimi trzema szczepami M1-podobnymi a podzbiorem genomów M1 o największym pokryciu (PmUG01, GN01, MA01, MOpte51017, MY01, UAN, THA05), oparta na 990 SNP z 45 kb sekwencji, możliwych do wywołania we wszystkich genomach. Łuki przerywane wskazują grupy pasożytów M1 i M1-podobne. Siatka w centrum sieci reprezentuje alternatywne ścieżki wprowadzone przez niezgodne SNP, na przykład w których Ptv_Leo i GA02 mają wspólny allel, a nie Ptv_Leo i GA01, jak w większości SNP. b Sieć filogenetyczna oparta na cechach wszystkich dostępnych genomów M1, w tym P. brasilianum i próbki pochodzącej od szympansa (MOpte51017), oparta na 2050 SNP z 1,1 Mb sekwencji. Oba klastry odpowiadają szczepom z Azji (klaster w lewym górnym rogu) i Afryki (klaster w prawym dolnym rogu). Klaster P. brasilianum i MOpte51017 pokrywają się z afrykańskimi szczepami ludzkimi. Podziałki wskazują substytucje/miejsca. Nazwy szczepów są oznaczone kolorami zgodnie z żywicielem lub pochodzeniem geograficznym, jak wskazano w legendzie.
Siatka w centrum sieci (Rys. 4a ) była zgodna z przepływem genów między różnymi liniami podobnymi do M1, ale niewiele wskazywało na to, że nastąpiła jakakolwiek wymiana między M1 a jakimkolwiek szczepem podobnym do M1 po radioterapii w obrębie kladu M1. Analiza głównych składowych (PCA) polimorfizmów na 1,5 Mb sekwencji w szczepach M1 o dużym pokryciu ( N = 16) i podobnych do M1 ( N = 2) była zgodna z tym wnioskiem, nie wykazując genomów M1 pośrednich między M1 i podobnymi do M1 (Rys. uzupełniający 4 ). Przeszukaliśmy również wszelkie regiony genomowe wykazujące niezwykle wysoką różnorodność w obrębie M1, gdzie niektóre szczepy mogą mieć niską dywergencję od podobnych do M1, potencjalnie odzwierciedlając niedawną introgresję ze szczepu podobnego do M1. Porównując każdą próbkę z genomem referencyjnym P. malariae , nanieśliśmy rozbieżne SNP na każde wywoływalne miejsce dla każdego szczepu w przesuwających się oknach wzdłuż każdego chromosomu; w przeciwieństwie do sieci filogenetycznej i PCA, podejście to pozwoliło na uwzględnienie regionów genomowych pokrytych tylko przez jeden lub dwa szczepy.
W prawie całym genomie (>99% z 20 kb okien w 22 Mb badanych), każdy szczep M1 różnił się od odniesienia o mniej niż 0,001 SNP na miejsce, podczas gdy trzy szczepy podobne do M1 (GA01, GA02 i Ptv_Leo) różniły się o ~0,005–0,015 SNP na miejsce (Rysunek uzupełniający 5 ). Znaleźliśmy tylko jeden region kandydacki, rozciągający się na około 45 kb na chromosomie 10, gdzie niektóre szczepy M1 były tak rozbieżne z odniesieniem jak GA01, podczas gdy GA02 wykazywał regiony, w których rozbieżność z odniesieniem była niezwykle niska (Rysunek 5a , Rysunek uzupełniający 6 ). W całym tym regionie żaden pojedynczy szczep nie był stale rozbieżny z odniesieniem lub stale podobny do GA02 (Rysunek uzupełniający 6 ). Tak więc, jeśli ten region odzwierciedla introgresję ze szczepu podobnego do M1, wydaje się, że następnie uległ rekombinacji z innymi szczepami M1. Zgodnie z tym, sieci filogenetyczne pochodzące z tego regionu wykazały prawie całkowitą siatkę, niezależnie od tego, czy opierały się tylko na małej liczbie szczepów, które obejmowały większość regionu (Rys. 5b ), czy na większej liczbie szczepów obejmujących mniej SNP (Rys. 5c , Ryc. uzupełniający 7 ). Region o dużej różnorodności rozciąga się na 12 genów, z których cztery wydają się być gospodarzami (prawdopodobnymi produktami są czynnik inicjacji translacji, czynnik ADP-rybozylacji, ligaza metioniny-tRNA i pseudourydynowa syntaza U2 snRNA/tRNA), podczas gdy dwa kodują białka o nieznanej funkcji konserwowane u P. falciparum i P. vivax , a pozostałe sześć jest niezidentyfikowanych i nie mają wyraźnych ortologów w innych gatunkach Plasmodium . Nie jest zatem jasne, jak ani dlaczego nowe allele mogły zostać selektywnie zachowane po introgresji. Odkrycia te sugerują, że w tym regionie chromosomu 10 zmienność wśród szczepów M1 pochodzących od człowieka odzwierciedla hybrydyzację ze szczepem podobnym do M1 pochodzącym od małp człekokształtnych, ale ponieważ region ten obejmuje jedynie około 0,2% badanej sekwencji genomu, introgresja wydaje się być bardzo rzadka.
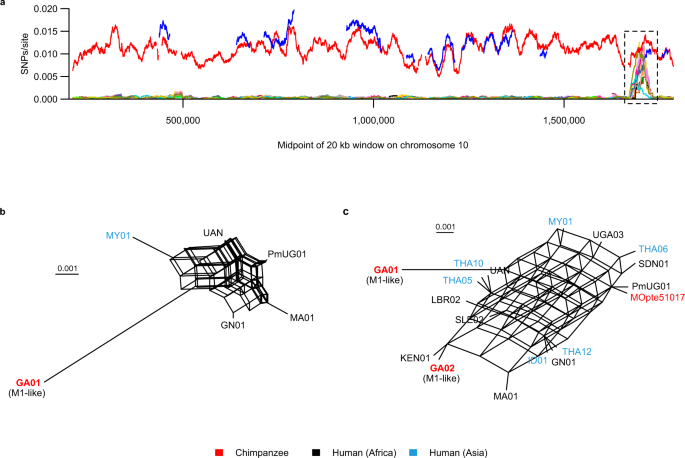
Dywergencja (SNP na miejsce możliwe do wywołania) z genomu referencyjnego M1 (PmUG01) jest przedstawiona na wykresie dla okien o długości 20 kb z krokiem 100 pb; wartości są przedstawione w punkcie środkowym każdego okna. Regiony subtelomerowe nie zostały przeanalizowane i nie są pokazane na wykresie, a miejsca o niskiej złożoności i miejsca niemożliwe do wywołania zostały zmienione na N. Uwzględniono tylko okna z co najmniej 40% zasad innych niż N, co doprowadziło do powstania luk w liniach. Szczepy podobne do M1 są przedstawione jako grube czerwone (GA01) i niebieskie (GA02) linie, a pozostałe cienkie kolorowe linie reprezentują szczepy M1. Szczep podobny do M1, Ptv_Leo, nie pojawia się na wykresie, ponieważ nie posiadał okien o długości 20 kb na tym chromosomie, które spełniałyby kryteria analizy. Czarne pole przerywane podkreśla region o wielkości około 45 kb (pozycje 1 680 100–1 723 901), w którym wiele szczepów M1 wykazuje niezwykle dużą dywergencję od odniesienia. Bardziej szczegółowy widok tego regionu pokazano na Rys. uzupełniającym 6. b Sieć filogenetyczna oparta na cechach wygenerowana z 415 SNP w regionie o wysokiej różnorodności, analizująca szczep GA01 podobny do M1 i pięć szczepów M1, które obejmowały większość regionu. Różnorodność nukleotydów parami wśród tych szczepów M1 wynosiła 0,0063 podstawień na miejsce. c Sieć filogenetyczna oparta na cechach wygenerowana z 26 SNP w regionie o wysokiej różnorodności, analizująca 16 szczepów M1 i dwa szczepy podobne do M1 o największym pokryciu genomu (Tabela uzupełniająca 4 ). Aby zostać uwzględnionym w sieciach, wymagane było, aby miejsce wariantu było objęte przez wszystkie szczepy, a wiele szczepów miało luki w pokryciu w różnych miejscach, dlatego dodanie większej liczby szczepów zazwyczaj zmniejszało liczbę analizowanych SNP. Nazwy szczepów w sieciach są oznaczone kolorami zgodnie z ich żywicielem lub pochodzeniem geograficznym, jak wskazano w legendzie. Podziałki wskazują 0,001 podstawień/miejsce.
Zmniejszona różnorodność i zwiększony polimorfizm niesynonimiczny w M1
Poprzednie badania porównujące gatunki ludzkiego Plasmodium (tj. P. falciparum i P. vivax ) z ich bliskimi krewnymi infekującymi małpy człekokształtne ujawniły różne poziomy i wzorce polimorfizmu nukleotydów u pasożytów od dwóch żywicieli11 , 12 , 36. W przypadku P. malariae sieć filogenetyczna (rys. 4a ) wskazywała, że linia podobna do M1 miała znacznie większą różnorodność niż M1, co jest zgodne z poprzednimi badaniami mniejszego zestawu danych24 . Obliczając średnią parami różnorodność nukleotydów (π), odkryliśmy, że zmienność genetyczna wśród trzech pasożytów podobnych do M1 od szympansów wynosiła 1,04%, prawie 40-krotnie więcej niż wartość (0,026%) obserwowana dla szczepów M1 (tabela 2 ). Aby zbadać możliwość, że większa różnorodność wśród szczepów podobnych do M1 była spowodowana zwiększoną częstością błędów sekwencjonowania, porównaliśmy liczbę zmian spowodowanych mutacjami tranzycyjnymi lub transwersyjnymi, ograniczając analizę do miejsc w sekwencjach kodujących, gdzie wszystkie możliwe zmiany są synonimiczne (miejsca czterokrotnie zdegenerowane). Błędy sekwencjonowania są znacznie częściej tranzycjami niż transwersjami37 , dlatego oczekuje się, że zbiór danych z większą liczbą błędów będzie miał wyższy stosunek tranzycji do transwersji niż zbiór z mniejszą liczbą błędów. Jednakże stosunek tranzycji do transwersji dla SNP w szczepach podobnych do M1 (1,25) był nieznacznie niższy niż w szczepach M1 (1,31), co wskazuje, że zwiększony błąd sekwencjonowania prawdopodobnie nie wyjaśnia znacznie większej różnorodności w szczepach podobnych do M1.
Różnica w zróżnicowaniu parzystym nukleotydów między szczepami M1 i M1-podobnymi była około 32-krotna w miejscach, w których wszystkie zmiany skutkują zmianą sekwencji aminokwasowej (miejsca zerokrotnie zdegenerowane), ale około 56-krotna w miejscach czterokrotnie zdegenerowanych (Tabela 2 ), co wskazuje, że M1 ma względny nadmiar zróżnicowania niesynonimicznego w porównaniu ze szczepem M1-podobnym. Obliczając stosunek polimorfizmów niesynonimicznych do synonimicznych dla każdej linii, odkryliśmy, że chociaż obie linie miały więcej polimorfizmów niesynonimicznych niż synonimicznych, stosunek (NS/S) był znacznie wyższy dla M1 (1,92) niż dla M1-podobnego (1,23). Wśród stałych różnic między szczepami M1 i podobnymi do M1, stosunek NS/S wynosił 1,24, co daje indeks neutralności wynoszący 38 (NI; obliczony poprzez podzielenie stosunku dla polimorfizmów przez wskaźnik dla stałych różnic) wynoszący 0,996, czyli bardzo blisko neutralnego oczekiwania na poziomie 1. Natomiast NI dla M1 wynosił 1,551, co ponownie wskazuje na nadmiar polimorfizmów niesynonimicznych i sugeruje, że to linia M1 jest pod tym względem nietypowa.
Chcieliśmy zbadać rozkład tego wzoru niesynonimicznego polimorfizmu w genach. Wiele genów w M1 nie miało synonimicznych polimorfizmów, a zatem nie można było zdefiniować wartości NI. Zamiast tego użyliśmy statystyki kierunku selekcji (DoS) 39 , która porównuje ułamek niesynonimicznych zmian wśród stałych różnic z ułamkiem wśród polimorfizmów i może być obliczona dla genów bez synonimicznych polimorfizmów. Wartości DoS dla szczepów podobnych do M1 były skoncentrowane wokół neutralnej wartości oczekiwanej 0, podczas gdy dla szczepów M1 cały rozkład został przesunięty w kierunku wartości ujemnych (rys. 6a ), co wskazuje, że większy ułamek polimorfizmów niż stałych różnic był niesynonimiczny. Zatem nadmiar niesynonimicznych polimorfizmów w M1 nie wynika z małej liczby nietypowych genów, ale jest powszechny w całym genomie.

a Wartości kierunku selekcji (DoS) dla M1 i M1-podobnych. Wykresy gęstości przedstawiają rozkład statystyki DoS w M1 (czerwony) i M1-podobnych (niebieski) dla 903 genów ze zdefiniowanymi wartościami DoS w obu liniach. Pionowe linie przerywane wskazują medianę wartości DoS dla każdej linii. b Widma częstości niesfałdowanych miejsc SNP w 1102 miejscach czterokrotnie (niebieski) i 4583 miejscach zerokrotnie (żółty) zdegenerowanych w M1.
Oczekuje się, że widmo częstotliwości miejsc (SFS) polimorfizmów będzie się różnić w zależności od poziomu selekcji, przy czym oczekuje się, że mutacje o silniejszym działaniu szkodliwym będą rzadsze. Zmiany synonimiczne są zwykle uznawane za neutralne, dlatego SFS polimorfizmów w miejscach czterokrotnie zdegenerowanych można wykorzystać jako przybliżenie neutralnego oczekiwania. Rozwinięty SFS dla polimorfizmów w miejscach zerokrotnie zdegenerowanych w szczepach M1 nie wykazał oczekiwanego nadmiaru tych niesynonimicznych zmian w klasie singletonów w porównaniu z polimorfizmami w miejscach czterokrotnie zdegenerowanych (ryc. 6b ). Wynik ten sugeruje, że te niesynonimiczne polimorfizmy w linii M1 były przedmiotem niewielkiej lub żadnej selekcji oczyszczającej.
Pochodzenie P. brasilianum
W naszych analizach mtDNA i loci jądrowych sekwencja P. brasilianum była identyczna z sekwencją P. malariae u ludzi (Rys. 1 ). Poprzednie badania dotyczące pochodzenia P. brasilianum podobnie wykorzystywały dane z jedynie niewielkiej liczby loci 4 , 24 , 25 , 26 , 29 , 30 , 31 . Aby zbadać tę kwestię bardziej szczegółowo, porównaliśmy genom P. brasilianum 40 z licznymi sekwencjami P. malariae , generując sieć filogenetyczną z 2050 SNP objętych wszystkimi szczepami M1 (Rys. 4b ). Sieć ta podzieliła ludzkie M1 (tj. P. malariae ) na dwa klastry, odpowiadające szczepom z Afryki i Azji, jak niedawno doniesiono 35 . Nie powinno dziwić, że próbka M1 pobrana od kameruńskiego szympansa w niewoli (MOpte51017) zgrupowała się z afrykańskimi szczepami P. malariae . Dodanie do tej sieci szczepu GA01, podobnego do M1, sugerowało, że korzeń M1 znajdował się blisko podstawy afrykańskiego skupiska (rys. uzupełniający 8 ), chociaż rozdzielczość w tym obszarze jest słaba, ponieważ w sieci dominują różnice między M1 a szczepami podobnymi do M1. P. brasilianum zgrupował się z afrykańskimi szczepami P. malariae (rys. 4b ), co sugeruje, że pochodził od szczepu ludzkiego zaimportowanego do Ameryki Południowej z Afryki.
Dyskusja
Wiadomo od około wieku, że szympansy są zarażane pasożytami morfologicznie nieodróżnialnymi od P. malariae u ludzi41 , 42. Pasożyt szympansa został nazwany P. rodhaini17 , ale kwestia, czy pasożyty małp człekokształtnych i ludzkich reprezentują dwa odrębne gatunki, była przedmiotem debaty, przy czym ostatecznie osiągnięto konsensus, że nie16 . W niniejszym artykule pokazujemy, że sekwencje spokrewnione z P. malariae z próbek małp człekokształtnych należą w rzeczywistości do dwóch zupełnie rozbieżnych gatunków i że oba zarażają goryle, a także szympansy. Spodziewamy się, że te dwa gatunki są morfologicznie podobne (tj. gatunki kryptyczne) i że oba mogły być obecne w oryginalnych badaniach mikroskopowych. Jeden z gatunków pasożytów małp człekokształtnych jest bardzo blisko spokrewniony z P. malariae , ale istnieje niewiele dowodów na niedawną wymianę genetyczną między nim a pasożytem ludzkim. Odkrycia te, wraz z obserwacjami dotyczącymi wzorców zróżnicowania genetycznego u pasożytów małp człekokształtnych i ludzi, pozwoliły nam w końcu rozwikłać pochodzenie P. malariae , a także pochodzenie P. brasilianum u małp Nowego Świata.
Charakterystyka molekularna gatunków pasożytów małp człekokształtnych bliżej spokrewnionych z P. malariae rozpoczęła się od sekwencji mtDNA nieco ponad 10 lat temu32 , 43 , 44 i została rozszerzona na prawie kompletne genomy jądrowe przez Rutledge’a i in.24 w 2017 roku, którzy nazwali te pasożyty P. malariae -like (powyżej użyliśmy M1-like). Rutledge i współpracownicy wyraźnie uważali, że P. malariae i P. malariae -like reprezentują dwa odrębne gatunki i rzeczywiście zasugerowali, że mogły się one oddzielić kilka milionów lat temu24 . Jednak wraz z charakterystyką trzeciego genomu P. malariae -like, nasze analizy wskazują na inny scenariusz: P. malariae mieści się w promieniu pasożytów P. malariae -like, nie różniąc się bardziej od pasożytów małp człekokształtnych niż one od siebie nawzajem (ryc. 4a ). Co więcej, fakt, że pasożyty ludzkie wykazują znacznie mniejszą zmienność genetyczną niż szczepy od małp człekokształtnych (Ryc. 4 ; Tabela 2 ) wskazuje, że P. malariae powstał w wyniku niedawnej transmisji pasożyta małp na ludzi. P. malariae wyraźnie doświadczył poważnego wąskiego gardła genetycznego, które najchętniej przypisuje się zdarzeniu transmisji międzygatunkowej. Zwiększony odsetek polimorfizmów w obrębie P. malariae , które są niesynonimiczne, w porównaniu z polimorfizmami w obrębie P. malariae -podobnymi, jest zgodny z szybką ekspansją populacji po tym wąskim gardle. Nasze skanowanie genomu rdzeniowego znalazło tylko jeden region, na chromosomie 10, gdzie różnorodność wśród szczepów P. malariae jest znacznie wyższa niż w innych częściach genomu. Region ten stanowi mniej niż 0,21% genomu rdzeniowego, co sugeruje, że nastąpiła bardzo niewielka introgresja z pasożytów małp człekokształtnych do populacji pasożytów ludzkich od czasu, gdy zaczęła się ona różnicować po swoim odzwierzęcym pochodzeniu.
Ta relacja między pasożytami związanymi z P. malariae u ludzi i małp jest zadziwiająco podobna do relacji między ludzkim P. falciparum a pasożytami małp z podrodzaju Laverania. P. falciparum wydaje się pochodzić od jednego gatunku pasożyta goryla ( P. praefalciparum ) całkiem niedawno, wykazuje znacznie zmniejszoną różnorodność genetyczną w porównaniu do pasożytów goryli i nie ma żadnych oznak introgresji z pasożytów goryli po powstaniu pasożyta ludzkiego 13 . Nie znaleziono żadnych dowodów na to, że P. praefalciparum lub inne gatunki Laverania zakażają ludzi żyjących w regionach bliskich zarażonym małpom 22 , 45 , 46 , tak jak nie stwierdzono zakażeń związanych z P. malariae pochodzących od małp u ludzi 22 .
Istotna różnica między dwoma kladami Plasmodium dotyczy ich specyficzności żywicieli. W naturze małpy człekokształtne Laverania wydają się być silnie specyficzne wobec żywicieli: trzy gatunki infekujące goryle nie zostały stwierdzone u dzikich szympansów, podczas gdy cztery gatunki infekujące szympansy i/lub bonobo (dwóch niezwykle blisko spokrewnionych żywicieli) nigdy nie zostały wykryte w próbkach pobranych od dzikich goryli36 . Podstawy tych barier dla transmisji międzygatunkowej Laverania są do pewnego stopnia poznane47 . Natomiast dwa gatunki pasożytów małp człekokształtnych spokrewnione z P. malariae zostały stwierdzone w próbkach pobranych zarówno od szympansów, jak i goryli, i nie wydaje się, aby a priori można było zakładać, że nie mogą one zarażać ludzi. Rzeczywiście, jakieś 70–80 lat temu donoszono, że P. malariae może być przenoszony na szympansy 48 i że „ P. rodhaini ” ( pasożyt spokrewniony z P. malariae obserwowany we krwi szympansów) może być przenoszony na ludzi 49 . Jednak większość tych eksperymentów obejmowała inokulację krwi zakażonej pasożytem, gdzie liczba przenoszonych pasożytów znacznie przekraczała liczbę tych z ukąszenia komara. Później Bray był w stanie zarazić komary Anopheles gambiae P. malariae od ludzi, którzy następnie mogli przenosić te pasożyty na szympansy 20 . Te obserwacje są zgodne z naszym odkryciem, że P. malariae pochodzenia ludzkiego zaraża małpy człekokształtne w niewoli. Natomiast Bray nie był w stanie zarazić A. gambiae pasożytami spokrewnionymi z P. malariae pochodzącymi od szympansów i doszedł do wniosku, że ryzyko odzwierzęcej transmisji pasożytów spokrewnionych z P. malariae z szympansów na ludzi jest znacznie niższe niż odwrotna antroponoza. Rzeczywiście, nasze analizy genomiczne wskazujące na rzadkość introgresji z pasożytów małp człekokształtnych do P. malariae dostarczają bezpośrednich dowodów na to, że transmisja odzwierzęca jest niezwykle rzadka i że P. malariae stała się w rzeczywistości nowym gatunkiem, wyizolowanym od spokrewnionych pasożytów małp człekokształtnych. Jeśli zostanie rozpoznany jako taki, P. malariae -like mógłby zachować nazwę P. rodhaini lub, biorąc pod uwagę, że pierwotny „ P. rodhaini ” był prawdopodobnie mieszanką dwóch rozbieżnych gatunków, mógłby zostać przemianowany na P. praemalariae , uznając jego rolę w powstaniu pasożytów ludzkich. Co ważne, dopóki nie poznamy podstaw pozornej izolacji pasożytów małp i ludzi, nie możemy wykluczyć możliwości, że P. praemalariae pojawi się ponownie u ludzi.
Od dawna wiadomo, że istnieje bardzo bliski związek między ludzkim P. malariae a P. brasilianum , pasożytem infekującym małpy Nowego Świata, ale kierunek transferu między gospodarzami był przedmiotem debaty, nawet całkiem niedawno 31 . Teraz, gdy wykazano , że P. malariae ma stosunkowo niedawne pochodzenie afrykańskie, jest jasne, że P. brasilianum powstał po wyeksportowaniu ludzkiego pasożyta do Ameryki Południowej, najprawdopodobniej w ciągu ostatnich 500 lat. Do tej pory scharakteryzowano tylko jeden genom P. brasilianum 40 , dlatego pozostaje niewiadome, czy te pasożyty, które, jak stwierdzono, zakażają około 30 różnych gatunków małp w kilku krajach Ameryki Południowej i Środkowej 50 , są wynikiem pojedynczego wprowadzenia, czy wielokrotnych transmisji od ludzi.
Istnienie drugiego pasożyta małp człekokształtnych spokrewnionego z P. malariae , nazwanego powyżej M2, zostało po raz pierwszy zasugerowane przez analizę sekwencji mtDNA (rys. 1a ; patrz również ref. 36 ). Złożyliśmy częściową sekwencję genomu M2 z odczytów, które pozostały niezauważone w wielokrotnie zakażonej próbce szympansa użytej do wygenerowania sekwencji genomu P. gaboni13 . Dane te wskazują, że zakres rozbieżności między M2 i P. malariae jest podobny do tego między P. vivax i jego krewnymi infekującymi małpy azjatyckie (np. P. knowlesi ) i większy niż wśród najbardziej rozbieżnych gatunków Laverania , takich jak P. falciparum i P. gaboni (rys. 3 ; tabela 1 ). Tak więc pasożyty M2 wyraźnie reprezentują wcześniej nieopisany gatunek, dla którego proponujemy nazwę Plasmodium celatum (w nawiązaniu do jego dotychczasowego ukrycia). Sekwencje tego pasożyta znaleziono w próbkach pobranych od szympansów, bonobo i goryli zachodnich, a także od komarów, przy czym ten ostatni gatunek, A. vinckei , okazał się być zarażony różnymi innymi pasożytami Plasmodium u szympansów i goryli 23 . Ponadto próbki M2-pozytywne zebrano w kilku miejscach w Kamerunie, Gabonie i Republice Konga, a także w jednym miejscu we wschodniej Demokratycznej Republice Konga. Zatem pasożyt ten jest szeroko rozpowszechniony i wydaje się nie wykazywać specyficzności żywiciela u małp człekokształtnych, chociaż nigdy nie został znaleziony u ludzi. Ponieważ dwa z trzech gatunków w linii pasożyta spokrewnionego z P. malariae zostały znalezione jako infekujące tylko afrykańskie małpy człekokształtne, podczas gdy trzeci odzwierciedla niedawną zoonozę, wydaje się jasne, że ta linia ewoluowała w Afryce i rzeczywiście wydaje się wysoce prawdopodobne, że całe promieniowanie pasożytów Plasmodium u ssaków miało tam swój początek 36 .
Skala czasowa pochodzenia gatunków Plasmodium infekujących ludzi była przedmiotem wielu spekulacji. Rutledge i współpracownicy oszacowali, że dywergencja P. malariae i P. malariae -like nastąpiła w tym samym czasie, co dywergencja, w obrębie Laverania , przodków P. falciparum i pasożyta szympansów P. reichenowi 24 . Zasugerowali, że te dwa wydarzenia w dwóch odrębnych liniach rozwojowych obejmowały przejście między naczelnymi innymi niż człowiek a ludźmi, a ich podobny czas może wskazywać, że oba były wynikiem wspólnego, leżącego u podstaw wydarzenia historycznego 24 . Jednakże obecnie jest jasne, że żadne z tych wydarzeń dywergencji nie wiązało się z transmisją z małpy na człowieka. Rzeczywiście, było już wiadomo, że ta rozbieżność w Laverania obejmowała podział między przodkami P. reichenowi i P. praefalciparum , pasożyta goryli9 , 51 , i że transmisja z goryla na człowieka, która dała początek P. falciparum, nastąpiła znacznie później11 , 13. Podobnie , nasze analizy wskazują obecnie, że wspólny przodek P. malariae i jakikolwiek genom z P. malariae -like, jak analizowali Rutledge i in., w rzeczywistości odzwierciedla koalescencję alleli w P. malariae -like, i ponownie transfer z małpy na człowieka, dający początek P. malariae, nastąpił znacznie później. Jedno z szacunków sugeruje, że P. falciparum powstał około 50 000 lat temu13 , 43 , chociaż zasugerowaliśmy bardziej współczesną oś czasu, 5000–10 000 lat temu36 . Niezależnie od wieku P. falciparum , pochodzenie P. malariae jest równie niedawne: poziom zróżnicowania genetycznego u P. malariae jest nawet niższy niż u P. falciparum36 , choć może to częściowo wynikać z wolniejszego tempa mutacji u pasożyta kwartanowego24 . Zatem mogło rzeczywiście mieć miejsce wspólne wydarzenie historyczne, które sprzyjało pojawieniu się zarówno P. falciparum, jak i P. malariae u ludzi, takie jak początki rolnictwa w Afryce.
Metody
Próbki DNA małp
Próbki małp zostały wybrane z istniejących banków okazów zebranych wcześniej od żyjących w niewoli i na wolności szympansów i goryli w zachodniej i centralnej Afryce (Tabela uzupełniająca 1 ). Pełną krew i wysuszone plamy krwi uzyskano od szympansów centralnych ( Pan troglodytes troglodytes) i nigeryjsko-kameruńskich ( P. t. ellioti ) przetrzymywanych w Centrum Ratowania Szympansów Sanaga Yong (SY) i Centrum Ratowania Dzikich Zwierząt Parku Narodowego Mfou (MO) w Kamerunie10 , 12. Ponadto niewielką ilość krwi (SAggg3157) uzyskano od zachodniego goryla nizinnego ( Gorilla gorilla gorilla ), który został zabity przez myśliwych i skonfiskowany w ramach programu antykłusowniczego kameruńskiego Ministerstwa Środowiska i Leśnictwa12. Pobrano próbki śledziony i płuc od dziko żyjącego, przyzwyczajonego szympansa zachodniego ( P. t. verus ) (Leo), który zmarł w 2002 roku w lesie Tai w Wybrzeżu Kości Słoniowej32. Na koniec pobrano próbki kału w sposób nieinwazyjny od małp człekokształtnych nieprzyzwyczajonych w licznych miejscach w Afryce równikowej 9 , 10 , 21 , 52 . DNA wyekstrahowano z krwi pełnej i wysuszonych plam krwi przy użyciu zestawu QIAamp Blood DNA Mini Kit lub Puregene Core Blood Kit (Qiagen). DNA z kału wyekstrahowano przy użyciu zestawu QIAamp Stool DNA mini kit (Qiagen). Wszystkie okazy poddano analizie mtDNA żywiciela w celu potwierdzenia ich pochodzenia gatunkowego i podgatunkowego. Pobieranie próbek w tamtym czasie zostało zatwierdzone przez Ministerstwa Środowiska i Leśnictwa poszczególnych krajów, a wszystkie próbki zostały wysłane zgodnie z przepisami Konwencji o międzynarodowym handlu dzikimi zwierzętami i roślinami gatunków zagrożonych wyginięciem oraz zezwoleniami na import i eksport obowiązującymi w poszczególnych krajach.
Diagnostyczna PCR w kierunku pasożytów związanych z P. malariae
Próbki krwi i kału małp zostały przebadane pod kątem sekwencji związanych z P. malariae metodą diagnostycznej PCR przy użyciu zestawów starterów specyficznych dla całego Plasmodium i P. malariae , jak opisano wcześniej9,21 . W skrócie, fragment mitochondrialnego cytB o długości 956 pb został zamplifikowany przy użyciu starterów specyficznych dla całego Plasmodium , a fragment mitochondrialnego cytB o długości 600 pb został zamplifikowany przy użyciu starterów specyficznych dla P. malariae (Tabela uzupełniająca 5 ). Amplikony zostały sekwencjonowane bez klonowania w międzyczasie i poddane analizie filogenetycznej w celu określenia gatunku zamplifikowanych sekwencji Plasmodium . Wcześniej doniesiono, że próbka śledziony szympansa Ptv_Leo jest pozytywna dla P. malariae32.
PCR z rozcieńczeniem granicznym
Próbki małp poddano SGA w celu uzyskania sekwencji Plasmodium bez artefaktów wywołanych PCR9 , 10 , 12 , 21 , 52. Próbka DNA została rozcieńczona w punkcie końcowym na płytkach 96-dołkowych, a rozcieńczenie, które dało mniej niż 30% dodatnich dołków, zostało użyte do wygenerowania sekwencji uzyskanych z pojedynczego matrycy. W przypadku próbek dodatnich dla P. malariae , regiony genów mitochondrialnych ( cytB i 2,5 kb cox1/cytB ) i jądrowych ( ldh, asl ) zostały amplifikowane przy użyciu zestawów starterów specyficznych dla P. malariae (Tabela uzupełniająca 5 , Rys. uzupełniający 1 ) i wcześniej opisanych warunków amplifikacji9 , 10 , 21 .
Selektywna amplifikacja całego genomu
Prawie pełnej długości genom M1 i częściowy genom podobny do M1 (odpowiednio) zostały amplifikowane z próbek krwi szympansa (MOpte51017) i śledziony (Ptv_Leo) przy użyciu SWGA, jak opisano wcześniej11 , 12. Krótko mówiąc, użyto niestandardowych skryptów do identyfikacji motywów sekwencji (6–12 pb), które często występowały w genomie referencyjnym P. malariae24 , ale tylko rzadko w genomie ludzkim. Zostały one przefiltrowane w celu usunięcia starterów, które wykazywały ekstremalne temperatury topnienia, przewidywano, że utworzą homodimery lub wiązały genom mitochondrialny P. malariae lub jego regiony subtelomerowe więcej niż trzy razy. Powstałe zestawy starterów, Pm_set37 i Pm_set31 (Tabela uzupełniająca 6 ), wykazały wysoką częstość występowania w genomie P. malariae i niską częstość występowania w genomie ludzkim. Reakcję SWGA przeprowadzono poprzez amplifikację DNA z krwi pełnej lub tkanek (100–750 ng) w 50 μl reakcji z buforem 1× phi29, 1 mM dNTP, 3,5 μM starterów SWGA (ekwimolarna mieszanina starterów w zestawie), 1% BSA i 30 jednostkami polimerazy phi29 (New England Biolabs). Warunki reakcji SWGA obejmowały 1-godzinny etap obniżania temperatury (35–30°C), a następnie 16-godzinny etap amplifikacji w temperaturze 30°C, po którym następowała 10-minutowa denaturacja phi29 w temperaturze 65°C. Produkty SWGA oczyszczono za pomocą kulek AMPure Beads (Beckman Coulter) i przechowywano w temperaturze 4°C. Aby uzyskać wystarczającą ilość genomowego DNA pasożyta do sekwencjonowania, próbki DNA pochodzące od małp człekokształtnych poddano wielokrotnym rundom (do 4) kolejnych amplifikacji SWGA, z których niektóre przeprowadzono z naprzemiennymi zestawami starterów w celu lepszego pokrycia genomu. Połączone produkty SWGA poddano sekwencjonowaniu MiSeq, a odczyty MiSeq przycięto za pomocą Cutadapt 53 w celu usunięcia sekwencji adaptorowej.
Eksploracja danych i montaż genomu M2
Pełne szczegóły metod bioinformatycznych użytych w tej sekcji i następnej podano w Uwadze Uzupełniającej 2. W skrócie, opublikowane biblioteki odczytów 13 , 15 zostały przefiltrowane w celu usunięcia odczytów gospodarza przez mapowanie na genom referencyjny gospodarza przy użyciu bwa 54 , a następnie mapowane na połączony odnośnik Plasmodium z bwa, po czym nastąpiło bardziej rygorystyczne mapowanie przy użyciu smalt ( www.sanger.ac.uk/science/tools/smalt-0 ; -y 0,6, tj. 60% zasad musiało być identycznych z odnośnikiem). Pary odczytów z obydwoma parami zmapowanymi na genom referencyjny P. malariae PmUG01 uznano za odczyty „ związane z P. malariae ” i złożono de novo przy użyciu SPades 55 . Konigi de novo o długości co najmniej 500 pb i pokryciu k-merów co najmniej 2X wyrównano do genomu podobnego do M1, PmlGA0124 , przy użyciu programu MUMmer 56 , a kontigi, które nie zostały zmapowane, wyrównano do PmUG01. Ponadto każdy zestaw odczytów powiązany z P. malariae wyrównano do istniejących sekwencji M2 dla asl i ldh przy użyciu programu smalt (-y 0,9, tj. 90% zasad musiało być identycznych z referencyjnymi), generując jedną sekwencję asl (numer 42) użytą na ryc. 1c .
Sekwencję pochodzącą z próbki PGABG03 ( ERS333073 13 ) wykorzystano do wygenerowania zestawu M2. Konigi M2 zidentyfikowano na podstawie podobieństwa do PmlGA01 lub PmUG01: kontigi o 83–92% identyczności nukleotydów z dowolnym odniesieniem uznano za M2, a kontigi o mniej niż 83% lub 92–97% identyczności nukleotydów sprawdzono ręcznie, aby ocenić, czy prawdopodobnie pochodzą z M2. Rusztowania pseudochromosomów wygenerowano z kontigów M2, układając je względem PmUG01 24 przy użyciu programu ABACAS 57. Aby ograniczyć błędy montażu w pobliżu końców kontigów, z końca każdego kontigu usunięto 100 pb, a następnie przeprowadzono ponowne wydłużenie za pomocą programu IMAGE 58 . Wykonano dwie kontrole w celu wykrycia błędnych złożeń, w tym błędnego włączenia sekwencji podobnej do M1: sprawdzono dopasowanie odczytów związanych z P. malariae do początkowego złożenia M2, aby zidentyfikować kontigi, dla których w zmapowanych odczytach reprezentowane były różne haplotypy; sprawdzono także dopasowania blastn złożenia do PmUG01 i PmlGA01, aby zidentyfikować kontigi z segmentami, które wykazywały nietypowo wysoką identyczność z PmlGA01 lub które były zmapowane na różne regiony genomów referencyjnych.
Sekwencje kodujące w końcowym zestawie M2 zostały adnotowane poprzez transfer z PmUG01 za pomocą RATT 59, a następnie poddane ręcznej inspekcji i selekcji. Skorygowane odległości genetyczne obliczono w programie R 60 (model TN93) na podstawie dopasowania nukleotydów każdego genu z M2 z jego ortologami z PmUG01 i szczepu GA01 podobnego do M1. Dopasowania aminokwasów wygenerowano dla genów, które miały ortologi jeden do jednego we wszystkich analizowanych gatunkach Plasmodium (ryc. 3 ), i wykorzystano je do określenia liczby różnic między parami gatunków, a tym samym do obliczenia odległości międzygatunkowych.
Porównanie szczepów M1 i podobnych do M1
Biblioteki odczytów pochodzące od szympansów (Ptv_Leo, MOpte51017, GA01 i GA02 24 ) zostały przefiltrowane w celu usunięcia odczytów sekwencyjnych z gospodarza i z koinfekcji innymi gatunkami Plasmodium przez mapowanie z bwa do połączonego odniesienia szympansa i Plasmodium . Pary odczytów wyrównane do P. malariae zostały użyte do wywołania wariantów, w połączeniu z bibliotekami odczytów M1 pochodzącymi od ludzi z trzech ostatnich badań 24 , 34 , 35 . Warianty z odniesienia (PmUG01) zostały wywołane i przefiltrowane przy użyciu GATK 61 z HaplotypeCaller, z wyłączeniem subtelomerów (Tabela uzupełniająca 7 ), regionów o niskiej złożoności (zidentyfikowanych przy użyciu Dustmasker 62 ) i miejsc, w których pojedyncza próbka miała więcej niż jeden alternatywny allel. W przypadku heterozygotycznego SNP, genotyp próbki uznano za allel potwierdzony przez większość odczytów. Pokrycie genomu referencyjnego dla każdego genomu (Tabela uzupełniająca 4 ) obliczono za pomocą GATK z wykorzystaniem CallableLoci, uznając za możliwe do wywołania miejsca objęte co najmniej pięcioma odczytami. SNP w genomie P. brasilianum 40 zidentyfikowano za pomocą MUMmer 56 poprzez porównanie złożonych kontigów z referencyjnym P. malariae i identyfikację różnic, ponieważ surowe dane sekwencjonowania dla tego izolatu nie były dostępne.
Główne składniki obliczono dla wariantów w genomach M1 i podobnych do M1 o największym pokryciu (tabela uzupełniająca 4 ) za pomocą programu plink 63. Widma częstości niesfałdowanych miejsc M1 uzyskano dla miejsc czterokrotnie lub zerokrotnie zdegenerowanych w PmUG01 za pomocą narzędzia est-sfs folder 64 , przy czym szczep GA01, podobny do M1, był grupą zewnętrzną. Aby zidentyfikować regiony o dużej różnorodności lub dywergencji, obliczono SNP/wywoływalną zasadę dla każdego szczepu M1 i podobnego do M1 w przesuwnych oknach o wielkości 20 kb w genomie referencyjnym z krokiem 100 pb, z wyłączeniem okien o liczbie wywoływalnych zasad mniejszej niż 8000.
Aby zbadać polimorfizm, wygenerowano sekwencje genów dla każdego szczepu, zmieniając sekwencję referencyjną na allel alternatywny w miejscach wariantowych lub na N w miejscach niemożliwych do odtworzenia, a następnie odrzucając pojedyncze sekwencje genów o niskim pokryciu referencyjnym. Pozostałe sekwencje, w tym PmUG01, wykorzystano do wygenerowania wyrównań nukleotydów dla niesubtelomerycznych i niepseudogennych. Polimorfizmy synonimiczne i niesynonimiczne oraz stałe różnice policzono, określając wpływ zmiany na PmUG01 lub GA01. Aby obliczyć średnie zróżnicowanie nukleotydów parami, wykluczono pozycje niemożliwe do odtworzenia w żadnym szczepie, a zróżnicowanie obliczono w R 60 dla wszystkich pozostałych miejsc oraz dla miejsc, które były czterokrotnie i zerokrotnie zdegenerowane w PmUG01.
Analizy filogenetyczne
Sekwencje nukleotydowe wyrównano za pomocą Clustal Omega 65 (rys. 1 ) lub TranslatorX/MUSCLE 66 (wszystkie pozostałe analizy) i przycięto, aby uwzględnić tylko miejsca obecne we wszystkich próbkach. Sekwencje aminokwasowe wyrównano za pomocą MUSCLE 67 i oczyszczono za pomocą Gblocks 68. Drzewa filogenetyczne skonstruowano za pomocą algorytmu RAxML 69 a, z 1000 powtórzeniami bootstrap, używając modelu GTR-Γ (do wyrównań nukleotydów) lub modelu PROTGAMMAJTTF (do wyrównań aminokwasów). Sieci filogenetyczne wygenerowano za pomocą SplitsTree4 70 z transformacją MedianNetwork, używając miejsc wariantowych zidentyfikowanych przez wywołanie SNP i wykluczając miejsca, które nie były bialleliczne lub były niemożliwe do wywołania w podzbiorze szczepów użytych w każdej sieci. Aby przeliczyć liczbę zmian na zmiany na miejsce, ilość sekwencji możliwych do wywołania we wszystkich szczepach w zestawie sekwencji została obliczona na podstawie plików wyjściowych CallableLoci przy użyciu narzędzi Bedtools 71 merge i Genomecov.
Podsumowanie raportu
Więcej informacji na temat projektu badawczego można znaleźć w podsumowaniu sprawozdawczości badań w czasopiśmie Nature Research Reporting Summary, do którego link znajduje się w tym artykule.
Dostępność danych
Nowe dane sekwencyjne podane w tym artykule zostały złożone w odpowiednich bazach danych NCBI pod numerami akcesyjnymi MN175636-MN175639 , MZ555468-MZ555486 , MZ927539 (GenBank, sekwencje SGA) i PRJNA767638 (Sequence Read Archive, sekwencje SWGA), jak wskazano w tabeli uzupełniającej 1. Rusztowania z nowego zestawu M2 pochodzącego z biblioteki sekwencji ERS333073 są dostępne w sekcji Third Party Annotation Section w bazach danych DDBJ/ENA/GenBank pod numerami akcesyjnymi TPA: BK061131-BK061144 . W badaniu przeanalizowano również publicznie dostępne dane z BioProjects PRJEB14392 24 i PRJNA344798 40 (zestawy genomów) oraz BioProjects PRJEB13344 24 , PRJEB12680 34 i PRJEB37746 35 (biblioteki odczytu), szczegóły w tabeli uzupełniającej 4 .
Odniesienia
-
Sutherland, CJ i Polley, SD W: Genetyka i ewolucja chorób zakaźnych (red. Tibayrenc, M.) 487–507 (Elsevier, 2017).
-
Sutherland, CJ Uporczywe pasożytnictwo: biologia adaptacyjna malarii wywoływanej przez malarię (Malarie) i malarii wywoływanej przez malarię owalną (Ovale). Trends Parasitol. 32 , 808–819 (2016).
-
Mueller, I., Zimmerman, PA i Reeder, JC Plasmodium malariae i Plasmodium ovale – „wstydliwe” pasożyty malarii. Trends Parasitol. 23 , 278–283 (2007).
-
Collins, WE i Jeffery, GM Plasmodium malariae : pasożyt i choroba. Clin. Microbiol. Rev. 20 , 579–592 (2007).
-
McKenzie, FE i Bossert, WH Zakażenia wielogatunkowe Plasmodium u ludzi. J. Parasitol. 85 , 12–18 (1999).
-
Oriero, EC, Amenga-Etego, L., Ishengoma, DS i Amambua-Ngwa, A. Plasmodium malariae , obecna wiedza i przyszłe możliwości badawcze dotyczące zaniedbanego gatunku pasożyta malarii. Crit. Rev. Microbiol. 47 , 44–56 (2021).
-
Scopel, KKG, Fontes, CJF, Nunes, Á. C., Horta, MF i Braga, É. M. Wysoka częstość występowania infekcji Plamodium malariae na endemicznym obszarze brazylijskiej Amazonii (Apiacás – stan Mato Grosso), wykryta za pomocą reakcji łańcuchowej polimerazy. Acta Trop. 90 , 61–64 (2004).
-
Mayxay, M., Pukrittayakamee, S., Newton, PN i White, NJ Zakażenia malarią mieszaną u ludzi. Trends Parasitol. 20 , 233–240 (2004).
-
Liu, W. i in. Pochodzenie pasożyta malarii ludzkiej Plasmodium falciparum u goryli. Nature 467 , 420–425 (2010).
-
Liu, W. i in. Afrykańskie pochodzenie pasożyta malarii Plasmodium vivax . Nat. Commun . 5 , 3346 (2014).
-
Sundararaman, SA i in. Genomy kryptycznych gatunków szympansów Plasmodium ujawniają kluczowe wydarzenia ewolucyjne prowadzące do malarii u ludzi. Nat. Commun. 7 , 11078 (2016).
-
Loy, DE i in. Historia ewolucji ludzkiego Plasmodium vivax ujawniona w analizach genomu spokrewnionych pasożytów małp człekokształtnych. Proc. Natl Acad. Sci. USA 115 , E8450–E8459 (2018).
-
Otto, TD i in. Genomy wszystkich znanych członków podrodzaju Plasmodium ujawniają ścieżki prowadzące do zjadliwej ludzkiej malarii. Nat. Microbiol. 3 , 687–697 (2018).
-
Prugnolle, F. i in. Różnorodność, zmiana żywicieli i ewolucja Plasmodium vivax infekującego afrykańskie małpy człekokształtne. Proc. Natl Acad. Sci. USA 110 , 8123–8128 (2013).
-
Gilabert, A. i in. Sekwencje genomu przypominające Plasmodium vivax rzucają nowe światło na biologię i ewolucję Plasmodium vivax . PLoS Biol. 16 , e2006035 (2018).
-
Coatney, GR, Collins, WE, Warren, M. i Contacos, PG Malaria u naczelnych (wydanie rządu USA, 1971).
-
Brumpt, E. Les parasites du paludisme des szympansy. CR Soc. Biol. 130 , 837–840 (1939).
-
Rodhain, J. & Dellaert, R. L’infection à Plasmodium malariae du szympans chez l’homme. Etude d’une premiere souche isolée de l’anthropoide Pan satyrus verus . Anna. Towarzystwo Belgia. Med. Trop. 23 , 19–46 (1943).
-
Rodhain, J. Les plasmodiums des anthropoïdes de l’Afrique centrale et leurs relations avec les plasmodiums humains. Byk. Acad. R. Med. Belgique 6 , 21–60 (1941).
-
Bray, RS Badania nad malarią u szympansów: VIII. Eksperymentalna transmisja i faza przederytrocytowa Plasmodium malariae , z uwagą na temat zasięgu żywicieli pasożyta. Am. J. Trop. Med. Hyg. 9 , 455–465 (1960).
-
Liu, W. i in. Dzikie bonobo są żywicielami pasożytów malarii występujących na ograniczonych obszarach geograficznych, w tym przypuszczalnie nowego gatunku Laverania . Nat. Commun. 8 , 1635 (2017).
-
Loy, DE i in. Badanie barier zakażeń odzwierzęcych dla pasożytów Plasmodium u małp człekokształtnych z wykorzystaniem analizy DNA w kale. Int. J. Parasitol. 48 , 531–542 (2018).
-
Makanga, B. i in. Przenoszenie malarii przez małpy człekokształtne i potencjalne przeniesienie z małpy na człowieka w. Afr. Proc. Natl Acad. Sci. USA 113 , 5329–5334 (2016).
-
Rutledge, GG i in. Genomy Plasmodium malariae i P. ovale dostarczają wglądu w ewolucję pasożyta malarii. Nature 542 , 101–104 (2017).
-
Fandeur, T., Volney, B., Peneau, C. & Thoisy, BD Małpy z lasów deszczowych Gujany Francuskiej są naturalnymi rezerwuarami malarii P. brasilianum / P. malariae . Parasitology 120 , 11–21 (2000).
-
Lalremruata, A. i in. Naturalne zakażenie Plasmodium brasilianum u ludzi: człowiek i małpa dzielą cztery pasożyty malarii w wenezuelskiej Amazonii. EBioMedicine 2 , 1186–1192 (2015).
-
Geiman, QM i Siddiqui, WA Wrażliwość małp Nowego Świata na malarię wywołaną przez człowieka. Am. J. Trop. Med. Hyg. 18 , 351–354 (1969).
-
Rayner, JC Malaria wywołana przez Plasmodium malariae : od małpy do człowieka? EBioMedicine 2 , 1023–1024 (2015).
-
Tazi, L. & Ayala, FJ Nieznany kierunek transferu gospodarza Plasmodium vivax v. P. simium i P. malariae v. P. brasilianum . Infect. Genet. Evol. 11 , 209–221 (2011).
-
Guimarães, LO i in. Różnorodność genetyczna Plasmodium malariae i Plasmodium brasilianum u żywicieli ludzkich, małpich i komarów w Brazylii. Acta Trop. 124 , 27–32 (2012).
-
Guimarães, LO i in. Różnorodność genetyczna białka powierzchniowego merozoitów-1 u Plasmodium malariae i Plasmodium brasilianum z Brazylii. BMC Infect. Dis. 15 , 529 (2015).
-
Kaiser, M. i in. Dzikie szympansy zakażone 5 gatunkami Plasmodium . Emerg. Infect. Dis. 16 , 1956–1959 (2010).
-
Leichty, AR i Brisson, D. Selektywna amplifikacja całego genomu w celu resekwencjonowania docelowych gatunków drobnoustrojów ze złożonych próbek naturalnych. Genetics 198 , 473–481 (2014).
-
Ansari, HR i in. Porównanie rozszerzonych rodzin genów w Plasmodium ovale wallikeri i Plasmodium ovale curtisi z Plasmodium malariae i innymi gatunkami Plasmodium w skali genomu . Int. J. Parasitol. 46 , 685–696 (2016).
-
Ibrahim, A. i in. Selektywna amplifikacja całego genomu DNA Plasmodium malariae z próbek klinicznych pozwala na wgląd w strukturę populacji. Sci. Rep. 10 , 10832 (2020).
-
Sharp, PM, Plenderleith, LJ i Hahn, BH Pochodzenie malarii u ludzi u małp. Annu. Rev. Microbiol. 74 , 39–63 (2020).
-
Hadigol, M. i Khiabanian, H. MERIT ujawnia wpływ kontekstu genomicznego na wskaźnik błędów sekwencjonowania w zastosowaniach ultragłębokich. BMC Bioinform. 19 , 219 (2018).
-
Rand, DM i Kann, LM Nadmiar polimorfizmu aminokwasów w DNA mitochondrialnym: kontrasty między genami Drosophila , myszy i ludzi. Mol. Biol. Evol. 13 , 735–748 (1996).
-
Stoletzki, N. & Eyre-Walker, A. Oszacowanie wskaźnika neutralności. Mol. Biol. Evol. 28 , 63–70 (2011).
-
Talundzic, E. i in. Pierwsza pełna sekwencja genomu Plasmodium brasilianum . Genome Announc. 5 , e01566–16 (2017).
-
Reichenow, E. Über das Vorkommen der Malariaparasiten des Menschen bei den Afrikanischen Menschenaffen. Centralny bl. F. Bakt. I. Abt. Oryg. 85 , 207–216 (1920).
-
Blacklock, B. i Adler, S. Pasożyt przypominający Plasmodium falciparum u szympansa. Ann. Trop. Med. Parasitol. XVI , 99–106 (1922).
-
Hayakawa, T. i in. Identyfikacja Plasmodium malariae , pasożyta wywołującego malarię u ludzi, u importowanych szympansów. PLoS ONE 4 , e7412 (2009).
-
Krief, S. i in. O różnorodności pasożytów malarii u małp afrykańskich i pochodzeniu Plasmodium falciparum od bonobo. PLoS Pathog. 6 , e1000765 (2010).
-
Sundararaman, SA i in. Pasożyty podobne do Plasmodium falciparum, infekujące dzikie małpy człekokształtne w południowym Kamerunie, nie stanowią nawracającego źródła malarii u ludzi. Proc. Natl Acad. Sci. USA 110 , 7020–7025 (2013).
-
Délicat-Loembet, L. i in. Brak dowodów na zakażenia malarią u ludzi w Gabonie. PLoS ONE 10 , e0126933 (2015).
-
Galaway, F., Yu, R., Constantinou, A., Prugnolle, F. i Wright, GJ Wskrzeszenie przodków ligandu inwazji RH5 dostarcza molekularnego wyjaśnienia pochodzenia malarii wywoływanej przez P. falciparum u ludzi. PLoS Biol. 17 , e3000490 (2019).
-
Rodhain, J. Wrażliwość szympansów na P. malariae pochodzenia ludzkiego. Am. J. Trop. Med. Hyg. s1-28 , 629–631 (1948).
-
Rodhain, J. Les plasmodiums des anthropoïdes de l’Afrique centrale et leurs relations avec les plasmodiums humains. Anna. Towarzystwo Belgia. Med. Trop. 20 , 489–505 (1940).
-
Deane, LM Malaria małpia w Brazylii. Mem. Inst. Oswaldo Cruz 87 , 1–20 (1992).
-
Rayner, JC, Liu, W., Peeters, M., Sharp, PM i Hahn, BH Mnogość gatunków Plasmodium u dzikich małp człekokształtnych: źródło zakażenia u ludzi? Trends Parasitol. 27 , 222–229 (2011).
-
Liu, W. i in. Wielogenomowa charakterystyka gatunków Plasmodium z podrodzaju Laverania zakażających dziko żyjące szympansy i goryle. Genome Biol. Evol. 8 , 1929–1939 (2016).
-
Martin, M. Cutadapt usuwa sekwencje adapterowe z odczytów sekwencjonowania o dużej przepustowości. EMBnet. J. 17 , 10–12 (2011).
-
Li, H. i Durbin, R. Szybkie i dokładne dopasowanie krótkich odczytów z transformacją Burrowsa–Wheelera. Bioinformatics 25 , 1754–1760 (2009).
-
Bankevich, A. i in. SPAdes: nowy algorytm składania genomu i jego zastosowania w sekwencjonowaniu pojedynczych komórek. J. Comput. Biol. 19 , 455–477 (2012).
-
Marçais, G. i in. MUMmer4: Szybki i wszechstronny system dopasowywania genomu. PLoS Comput. Biol. 14 , e1005944 (2018).
-
Assefa, S., Keane, TM, Otto, TD, Newbold, C. i Berriman, M. ABACAS: automatyczna koniguacja sekwencji zmontowanych oparta na algorytmie. Bioinformatics 25 , 1968–1969 (2009).
-
Tsai, IJ, Otto, TD i Berriman, M. Ulepszanie projektów montażowych poprzez iteracyjne mapowanie i montaż krótkich odczytów w celu wyeliminowania luk. Genome Biol. 11 , R41 (2010).
-
Otto, TD, Dillon, GP, Degrave, WS i Berriman, M. RATT: narzędzie do szybkiego transferu adnotacji. Nucleic Acids Res 39 , e57–e57 (2011).
-
Paradis, E., Claude, J. i Strimmer, K. APE: analiza filogenetyki i ewolucji w języku R. Bioinformatics 20 , 289–290 (2004).
-
Auwera, GA i in. Od danych FastQ do wywołań wariantów o wysokim poziomie ufności: najlepsze praktyki w zakresie zestawu narzędzi do analizy genomu. Curr. Protoc. Bioinform. 43 , 11.10.1–11.10.33 (2013).
-
Camacho, C. i in. BLAST+: architektura i zastosowania. BMC Bioinform. 10 , 421 (2009).
-
Chang, CC i in. Druga generacja PLINK: stawianie czoła wyzwaniom związanym z większymi i bogatszymi zbiorami danych. Gigascience 4 , 1–16 (2015).
-
Keightley, PD i Jackson, BC Wnioskowanie o prawdopodobieństwie stanu allelicznego pochodnego w porównaniu z przodkami w miejscu polimorficznym. Genetics 209 , 897–906 (2018).
-
Sievers, F. i in. Szybka, skalowalna generacja wysokiej jakości wielosekwencyjnych dopasowań białek przy użyciu Clustal Omega. Mol. Syst. Biol. 7 , 539 (2011).
-
Abascal, F., Zardoya, R. i Telford, MJ TranslatorX: wielokrotne dopasowanie sekwencji nukleotydowych na podstawie translacji aminokwasów. Nucleic Acids Res. 38 , W7–W13 (2010).
-
Edgar, RC MUSCLE: wielokrotne dopasowanie sekwencji z wysoką dokładnością i wysoką przepustowością. Nucleic Acids Res. 32 , 1792–1797 (2004).
-
Castresana, J. Selekcja konserwatywnych bloków z wielu dopasowań do wykorzystania w analizie filogenetycznej. Mol. Biol. Evol. 17 , 540–552 (2000).
-
Stamatakis, A. RAxML wersja 8: narzędzie do analizy filogenetycznej i postanalizy dużych filogenez. Bioinformatics 30 , 1312–1313 (2014).
-
Huson, DH Zastosowanie sieci filogenetycznych w badaniach ewolucyjnych. Mol. Biol. Evol. 23 , 254–267 (2005).
-
Quinlan, AR i Hall, IM BEDTools: elastyczny zestaw narzędzi do porównywania cech genomicznych. Bioinformatics 26 , 841–842 (2010).
Podziękowanie
Niniejsza praca została dofinansowana grantami od Narodowych Instytutów Zdrowia (NIH) R01 AI091595 (BHH i PMS), R01 AI120810 (BHH), R01 AI050529 (BHH) i P30 AI045008 (BHH). Dziękujemy Richardowi Carterowi za pomocne dyskusje oraz Thomasowi Otto za umożliwienie dostępu do pełnych bibliotek sekwencjonowania z GA01 i GA02.
Deklaracje etyczne
Konflikty interesów
Autorzy deklarują brak konfliktu interesów.
Recenzja ekspercka
Informacje o recenzjach eksperckich
Nature Communications pragnie podziękować anonimowemu recenzentowi/recenzentom za wkład w recenzję niniejszej pracy.
Informacje dodatkowe
Uwaga wydawcy: Springer Nature zachowuje neutralność w kwestii roszczeń jurysdykcyjnych zawartych w opublikowanych mapach oraz powiązań instytucjonalnych.
Informacje uzupełniające
Prawa i uprawnienia
Otwarty dostęp Niniejszy artykuł jest licencjonowany na podstawie licencji Creative Commons Uznanie autorstwa 4.0 Międzynarodowe, która zezwala na używanie, udostępnianie, adaptację, dystrybucję i reprodukcję na dowolnym nośniku lub w dowolnym formacie, pod warunkiem wskazania autorów i źródła, podania linku do licencji Creative Commons oraz wskazania, czy wprowadzono zmiany. Obrazy lub inne materiały stron trzecich w tym artykule są objęte licencją Creative Commons, o ile nie wskazano inaczej w informacji o autorstwie materiału. Jeśli materiał nie jest objęty licencją Creative Commons, a zamierzony sposób wykorzystania nie jest dozwolony przez przepisy ustawowe lub wykracza poza dozwolony zakres, należy uzyskać zgodę bezpośrednio od właściciela praw autorskich. Aby wyświetlić kopię tej licencji, odwiedź stronę http://creativecommons.org/licenses/by/4.0/ .
Komunikacja przyrodnicza tom 13 , Numer artykułu: 1868 ( 2022 )
Link do artykułu: https://www.nature.com/articles/s41467-022-29306-4

 / Piotr Kotlarz")
 / Piotr Kotlarz")

 / Piotr Kotlarz")

{kind=link}