


Ostatnia dekada przyniosła wiele sukcesów w identyfikacji genów ryzyka zaburzeń ze spektrum autyzmu (ASD), a wiele badań wskazywało na mutacje powodujące utratę funkcji (LoF) w obrębie tych genów. Mimo to nie poczyniono dotychczas żadnych znaczących postępów klinicznych w opracowywaniu metod leczenia ASD. Biorąc pod uwagę rolę mutacji LoF w etiologii ASD, wiele opracowywanych leków ma na celu ratowanie haploinsuficientnego działania genów na poziomach transkrypcji, translacji i białka. W tym przeglądzie omówione zostaną różne techniki terapeutyczne opracowywane na każdym poziomie centralnego dogmatu, na przykładach obejmujących: aktywację CRISPR (CRISPRa) i wymianę genów na poziomie DNA, oligonukleotydy antysensowne (ASO) na poziomie mRNA oraz leki małocząsteczkowe na poziomie poziom białka, a następnie przegląd aktualnych metod dostarczania tych leków. Ponieważ penetracja centralnego układu nerwowego (OUN) ma ogromne znaczenie w leczeniu ASD, szczególnie konieczna jest ocena metod podawania leku, które charakteryzują się większą skutecznością w przekraczaniu bariery krew-mózg (BBB).
Wstęp
Zaburzenia ze spektrum autyzmu (ASD) to zaburzenie neurorozwojowe (NDD), które charakteryzuje się trzema podstawowymi objawami: deficytami w interakcjach społecznych i komunikacji, rozwojem języka oraz restrykcyjnymi i powtarzalnymi zachowaniami. U dużej części dzieci, u których zdiagnozowano ASD, występują dodatkowe objawy, w tym deficyty poznawcze, opóźnienie rozwoju, stany lękowe i inne choroby współistniejące z zaburzeniami nastroju i zaburzeniami psychicznymi [ 1 , 2 ]. Według stanu na rok 2021 CDC szacuje, że u 1 na 44, czyli ~2,3% dzieci w Stanach Zjednoczonych, zdiagnozowano ASD [ 3 ].
Z szacunków wynika, że 70% osób z ASD ma ograniczoną zdolność do samodzielnego życia [ 4 ]. Ta trwająca całe życie zależność od opiekunów, a także związane z ASD deficyty społeczne, poznawcze i behawioralne mogą przyczyniać się do stresu rodziców, prowadząc do zwiększonej liczby rozwodów wśród rodziców dzieci, u których zdiagnozowano ASD [ 5 ]. Na poziomie finansowym przewidywany koszt zasobów potrzebnych do opieki nad osobami z ASD będzie stopniowo wzrastał do 5,54 biliona dolarów rocznie do 2060 r. ze względu na koszty edukacji specjalnej, utratę produktywności spowodowaną nieformalną opieką i zwiększone korzystanie z usług opieki zdrowotnej [ 6 ]. Biorąc pod uwagę powszechność diagnozy, stres rodzinny i obciążenie finansowe społeczeństwa, konieczne jest opracowanie i udoskonalenie technik łagodzenia objawów społecznych, poznawczych i behawioralnych w ASD.
Powszechnie wiadomo, że ASD ma silne podłoże genetyczne. Wcześniejsze badania wykazały, że u bliźniąt jednojajowych występuje znacznie większa zgodność pod względem ASD niż u bliźniąt dwuzygotycznych, a odziedziczalność ASD szacuje się na 83% [ 7 ]. Chociaż osoby z niektórymi przyczynami monogenowymi, takimi jak zespół Angelmana (AS), zespół łamliwego chromosomu X (FXS) i zespół Retta (RTT) [ 8 , 9 , 10 ], mają cechy ASD, etiologia ASD jako całości jest niezwykle heterogeniczny [ 11 , 12 ]. Wcześniejsze badania zidentyfikowały rzadkie de novo i odziedziczone warianty liczby kopii (CNV) jako główne czynniki zwiększające ryzyko ASD [ 13 , 14 , 15 , 16 , 17 , 18 , 19 ]. Następnie sekwencjonowanie całego egzonu (WES) rodzin prostych z jednym chorym dzieckiem wykazało silny związek rzadkich wariantów pojedynczych nukleotydów eksonowych de novo (SNV) z ASD [ 20 , 21 , 22 , 23 , 24 ], przy czym nowsze analizy podkreślają około stu genów ryzyka ASD istotnych dla całego genomu [ 25 , 26 ]. Dla podzbioru genów, które znacznie przekroczyły znaczącą wartość odcięcia dla całego genomu (co najmniej FDR < 0,05), takich jak KMT2E , ANKRD11 , ARID1B , CHD8 , PTEN, SHANK3, DYRK1A i CUL3 , modele mysie opracowano w ciągu lata [ 27 , 28 , 29 , 30 , 31 , 32 , 33 , 34 ]. Ponadto ustalono modele naczelnych innych niż ludzie (NHP) dla genów MECP2 (związanych z RTT) i SHANK3 u makaków cynomolgus ( Macaca fascicularis ), aby lepiej podsumować punkty czasowe rozwoju człowieka [ 35 , 36 ]. Modele te dodatkowo sugerują, że heterozygotyczne mutacje powodujące utratę funkcji (LoF) (znane również jako haploinsufficiency) w tych genach są odpowiedzialne za określone fenotypy neurobiologiczne i behawioralne zwierząt. Oprócz rzadkich wariantów de novo, w niedawnym badaniu asocjacyjnym obejmującym cały genom (GWAS) zidentyfikowano 5 loci ASD istotnych dla całego genomu [ 37] Biorąc pod uwagę tak ekstremalną heterogeniczność genetyczną i jednoznaczną rolę genów dotkniętych LoF (i często haploinsuficientnych) w etiologii ASD, bezcenne będzie odejście od badań opartych na identyfikacji i przystąpienie do badania technik terapeutycznych, które mogłyby zwiększyć ekspresję tych genów .
Interwencje terapeutyczne w ASD mające na celu ratowanie haploinsufficiency poszczególnych genów można opracować tak, aby były ukierunkowane na wszystkie trzy poziomy centralnego dogmatu biologii molekularnej: DNA, mRNA i białko ( ryc. 1 ) . Przykłady takich interwencji obejmują edycję genomu za pomocą CRISPR na poziomie genetycznym, oligonukleotydy antysensowne (ASO) zarówno na poziomie transkrypcyjnym, jak i potranskrypcyjnym, a także zastosowanie leków drobnocząsteczkowych do celowania w szlaki molekularne na poziomie translacji lub białka. W tym przeglądzie przeanalizujemy zalety i wady różnych technik w ramach głównego dogmatu w celu ratowania fenotypów związanych z ASD.

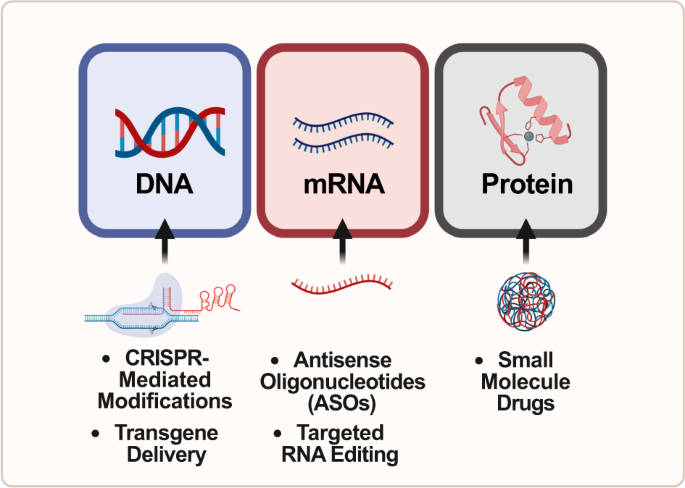
Na rysunku przedstawiono leki liniowo w oparciu o naturalny postęp ekspresji genów. W pierwszej kolumnie leki małocząsteczkowe i terapie oparte na CRISPR są zwykle opracowywane w celu leczenia chorób na poziomie DNA. W drugiej kolumnie oligonukleotydy antysensowne (ASO) są zwykle opracowywane w celu leczenia chorób na poziomie mRNA. W trzeciej kolumnie leki małocząsteczkowe są zwykle opracowywane w celu leczenia chorób na poziomie białek. W całym przeglądzie opisano konkretne przykłady każdego z tych środków terapeutycznych.
Ratunek na poziomie DNA
Terapia genowa obejmuje techniki, które mogą zmienić ekspresję genów organizmu poprzez celowanie w DNA poprzez dostarczenie transgenu lub bezpośrednią modyfikację genomu, w celu terapeutycznego przywrócenia normalnego poziomu ekspresji genu ulegającego patologicznej ekspresji [ 38 ].
Dostarczanie transgenu
Dostarczenie transgenu jest metodą terapii genowej, którą można zastosować w celu skorygowania haploinsuficiencji spowodowanej mutacjami LoF [ 39 ]. Zastosowano go w leczeniu kilku NDD, dla których znana jest przyczyna monogenowa. RTT to NDD mieszczące się w klasyfikacji ASD [ 40 , 41 ]. Podczas gdy LoF genu białka wiążącego metylo-CpG 2 ( MECP2 ) jest przyczyną RTT, duplikacja MECP2 powoduje zespół duplikacji MECP2 (MDS) [ 10 , 42 ]. Wykazano, że możliwe jest zmniejszenie ciężkości RTT poprzez dostarczenie transgenu Mecp2 w mysim modelu Mecp2-null [ 43 ]. Jednakże, biorąc pod uwagę czułość dawkowania genu MECP2 , przed translacją kliniczną konieczne jest właściwe określenie dawki. Nowsze badanie wykazało, że dostarczenie podatnej na niestabilność kasety transgenu Mecp2 ( iMecp2 ) przy użyciu wektora wirusa związanego z adenowirusem (AAV) objawowym myszom z mutacją Mecp2 znacząco poprawiło aktywność lokomotoryczną, długość życia i normalizację ekspresji genów [ 44 ].
Zespół łamliwego chromosomu X, kolejna NDD, która charakteryzuje się między innymi niepełnosprawnością intelektualną (ID), często współistnieje z ASD i jest wynikiem mutacji ekspansji powtórzeń tripletowych CGG w genie upośledzenia umysłowego łamliwego X 1 ( FMR1 ), która wycisza produkcję kodowanego przez niego białka FMRP [ 45 , 46 ]. W jednym badaniu wykorzystano dostarczanie transgenu nierozszerzonej kopii genu FMR1 przy użyciu wektora AAV wstrzykniętego bezpośrednio do mózgów myszy Fmr1 -/- , co skutecznie uratowało myszy o powtarzających się fenotypach behawioralnych, społecznych i napadowych [ 47 ]. Poza kontekstem NDD FDA zatwierdziła Luxturnę – terapię transgenową mającą na celu dostarczenie genu RPE65 do siatkówki i skuteczną terapię rzadkiej dziedzicznej choroby siatkówki, która powoduje zaburzenia widzenia i ślepotę [ 48 , 49 ]. Biorąc pod uwagę zgodę Luxturny i sukcesy w dostarczaniu transgenów dla modeli myszy RTT i FXS, mechanizm ten może mieć wartość dla innych genów LoF związanych z ASD. Należy jednak pamiętać, że terapeuci NDD mogą napotkać w klinice więcej przeszkód niż podawanie leku Luxturna do siatkówki, ponieważ mózg jest trudniej dostępny niż oko i wymaga, aby wektor dostarczania przekroczył barierę krew-mózg (BBB). Co więcej, mózg jest bardziej złożonym organem, charakteryzującym się większą złożonością typów komórek i znacznie różniącymi się poziomami ekspresji genów pomiędzy każdym typem, co sprawia, że specyficzność komórkowa stanowi dodatkowy problem w terapii NDD [ 50 ].
Modyfikacje za pośrednictwem CRISPR
W ciągu ostatniej dekady pojawienie się inżynierii genomu za pomocą CRISPR-Cas9 zrewolucjonizowało terapię genową, otwierając nową drogę terapeutyczną opartą na modyfikacjach na poziomie DNA. CRISPR-Cas9 to konstrukt składający się z kierującego RNA (gRNA), który celuje w interesujące loci genetyczne oraz enzymu endonukleazy Cas9, który działa jak para nożyczek nukleotydowych, które przecinają DNA w miejscu docelowym, skutecznie tworząc dwuniciowy Przerwa w DNA w celu późniejszej edycji genomu [ 51 ]. Wyjątkowa zdolność kierowania Cas9 do dowolnej lokalizacji w genomie otworzyła nowe możliwości celowanej terapii genowej.
Jedną z możliwości wykorzystania inżynierii genomu do regulacji ekspresji genów jest skierowanie jej na naturalne transkrypty antysensowne (NAT) [ 52 ]. NAT ulegają endogennej ekspresji zarówno w organizmach prokariotycznych, jak i eukariotycznych. W układach eukariotycznych NAT mogą wywierać dwukierunkowy wpływ regulacyjny na transkrypcję genów docelowych, albo tłumiąc, albo wzmacniając translację mRNA genu docelowego [ 53 , 54 ]. Hamowanie ekspresji docelowego genu może zachodzić poprzez różne mechanizmy, takie jak interferencja RNA po utworzeniu dupleksu sensownego-antysensownego mRNA, interferencja transkrypcyjna, w której NAT może działać jako fizyczna bariera dla aktywności polimerazy RNA lub epigenetyczna metylacja sensu genu DNA, hamując w ten sposób transkrypcję sensownego transkryptu mRNA [ 55 , 56 ]. Chociaż wiele NAT ma hamującą kontrolę nad ekspresją swoich komplementarnych genów, istnieją pewne przypadki, w których NAT mogą bezpośrednio zwiększać sensowną ekspresję genów [ 57 ]. Proponuje się, że NAT mogą zwiększać ekspresję docelowego genu poprzez zwiększanie stabilności sensownego mRNA lub modyfikacje epigenetyczne związane z euchromatyną [ 54 ]. Umieszczając to w kontekście terapeutycznym, możliwe może być przywrócenie ekspresji genów ryzyka ASD, które mają mutacje LoF, poprzez ukierunkowaną supresję odpowiednich hamujących NAT.
Strategię tę ostatnio zastosowano w leczeniu zespołu Angelmana (AS), choroby NDD, której przyczyną może być mutacja LoF w matczynej kopii allelu UBE3A [ 58 , 59 ]. Ponieważ ojcowska kopia UBE3A jest zwykle nieaktywna, badanie hamowania UBE3A NAT ma wartość terapeutyczną. Odnotowano sukces w ratowaniu haploinsufektywności genu UBE3A poprzez hamowanie transkrypcji UBE3A NAT za pośrednictwem CRISPR-Cas9 u myszy – skutecznie przywracając ekspresję UBE3A poprzez reaktywację kopii ojcowskiej [ 58 , 60 ]. W kontekście FXS, zamiast celować w NAT genu FMR1 , udało się przywrócić ekspresję FMR1 w indukowanych pluripotencjalnych komórkach macierzystych (iPSC) poprzez bezpośrednią delecję patologicznych sekwencji powtórzeń sensownego genu za pośrednictwem CRISPR-Cas9 [ 61 ] .
Poza kontekstem NDD, obecnie podejmuje się próby zastosowania CRISPR-Cas9 do leczenia wrodzonej ślepoty Lebera 10 (LCA10). LCA10 to ciężka postać dystrofii siatkówki spowodowana mutacją punktową adeniny do guaniny w intronie 26 ludzkiego genu CEP290 (IVS26), co skutkuje włączeniem tajemniczego egzonu i przedwczesnego kodonu stop [ 62 ]. Aby celować w LCA10, w jednym badaniu opracowano lek o nazwie EDIT-101, który składa się z Cas9 i konstruktu gRNA upakowanego w serotypie 5 AAV (AAV5) w celu wprowadzenia delecji lub inwersji w zmutowanym regionie intronu 26 [ 63 ]. W mysim modelu ludzkiego CEP290 IVS26, EDIT-101 wykazywał dużą wydajność edycji w komórkach fotoreceptorów, skutecznie przywracał prawidłowy splicing mRNA CEP290 i przywracał produkcję białka CEP290 pełnej długości [ 63 ]. Na poziomie klinicznym EDIT-101 jest pierwszym zastosowaniem edytora genomu w OUN i obecnie znajduje się w fazie 1/2 badań klinicznych z udziałem pacjentów dorosłych i dzieci ( https://clinicaltrials.gov/ct2/show/NCT03872479 ).
Zmodyfikowana aktywacja CRISPR
Wykorzystując jako podstawę tradycyjny kompleks CRISPR-Cas9, opracowano zmodyfikowaną wersję, w której enzym Cas9 jest nieaktywny lub „martwy” (dCas9) [ 64 ]. Chociaż zdolność dCas9 do cięcia za pośrednictwem endonukleazy staje się nieaktywna, enzym może nadal wiązać się, umożliwiając specyficzne ukierunkowanie w obrębie genomu. Ten system dCas9 można połączyć z aktywatorami transkrypcji (ryc. 2A ) w celu zwiększenia ekspresji genów bez wywoływania pęknięcia dwuniciowego DNA – procesu znanego jako aktywacja za pośrednictwem CRISPR (CRISPRa) [ 65 , 66 , 67 ] (Rys. 2B ). Aby CRISPRa zadziałała, RNA o pojedynczym przewodniku (sgRNA) zaprojektowano tak, aby celowało w regiony od 1 do 1000 bp powyżej miejsca startu transkrypcji (TSS) w regionie promotora genu docelowego. Gdy sgRNA zwiąże się z tym regionem, rekrutowany będzie dCas9 połączony z odpowiednimi aktywatorami transkrypcji.

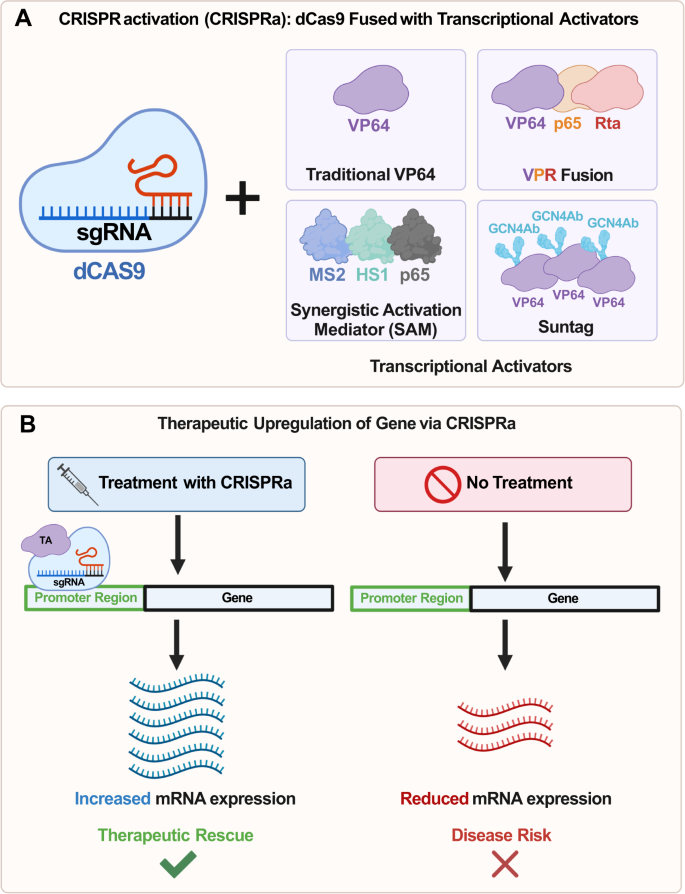
CRISPRa wykorzystuje katalitycznie martwy Cas9 (dCas9) połączony z różnymi aktywatorami transkrypcji.
Przedstawienie najpowszechniejszych aktywatorów transkrypcji (TA) połączonych z dCas9: (i) Tradycyjny VP64, (ii) Kombinacja VP64, p65 i Rta (VPR), (iii) System synergistycznego mediatora aktywacji (SAM) składający się p65, HSF1 i MS2 oraz (iv) system Suntag składający się z dCas9 połączonego z łańcuchem polipeptydowym, w którym fuzja przeciwciała VP64-GCN4 jest związana z każdym peptydem – umożliwiając rekrutację wielu kopii VP64. B Mechanizm działania CRISPRa. Po naprowadzeniu przez konstrukt sgRNA do regionu promotora, 1 do 1000 bp powyżej miejsca startu transkrypcji (TSS) genu docelowego, dCas9-VPR będzie sprzyjał wzrostowi transkrypcji. Zmniejszenie ekspresji mRNA w prawej kolumnie jest wynikiem nieleczonego genu Haploinsufficient. Skróty : Aktywator transkrypcji TA, Transaktywator Rta wirusa Epsteina-Barra R, MS2 – białko płaszcza bakteriofaga, ludzki czynnik szoku cieplnego 1 HSF1, przeciwciało GCN4Ab GCN4.
Możliwe jest modulowanie wielkości aktywacji poprzez zmianę, które z białek jest połączone z dCas9 (ryc. 2A ). Po fuzji z C-końcem dCas9 aktywator transkrypcji VP64 powoduje umiarkowany wzrost ekspresji genów [ 68 ]. Opierając się na tej początkowej fuzji VP64, kombinacyjna fuzja VP64, p65 i Rta (VPR) z C-końcem dCas9 powoduje znacznie wyższą aktywację transkrypcji, z wielkością efektu w zakresie od 22-krotnie do 320-krotnie większą niż sam VP64 [ 68 ] Stycznie, SunTag to kolejny system wykorzystujący VP64. Jednakże zamiast VP64 bezpośrednio połączonego z dCas9, Suntag składa się z dCas9 połączonego z łańcuchem polipeptydowym, w którym fuzja przeciwciała VP64-GCN4 jest związana z każdym peptydem, co pozwala na rekrutację wielu kopii VP64 do promotora genu [ 69 ] . Inną fuzją dCas9 jest system synergistycznego mediatora aktywacji (SAM), który składa się łącznie z trzech składników: dCas9 połączony z VP64 z sekwencją lokalizacji jądrowej (NLS) (NLS-dCas9-VP64), dwie domeny aktywacyjne (p65 i ludzki szok cieplny czynnik 1 (HSF1)) połączony z białkiem płaszcza bakteriofaga MS2 (MS2-p65-HSF1) i sgRNA połączony z dwoma aptamerami spinki do włosów nakierowanymi na białko MS2 w regionach łodyga-pętla 2 i tetraloop (sgRNA2.0) [ 70 ] .
Dla porównania, jedno badanie wykazało, że chociaż systemy VPR, Suntag i SAM wykazywały o wiele rzędów wielkości większą aktywację transkrypcji niż sam VP64, różnice między tymi trzema systemami wynosiły nie więcej niż jeden poziom [ 71 ]. Chociaż należy zauważyć, że różnice między VPR, Suntag i SAM zależą od typu komórki i są specyficzne dla genu. Co więcej, wszechobecne stosowanie silnego aktywatora transkrypcji w CRISPRa może nie być korzystne, ponieważ nadmierna aktywacja niektórych genów może wywołać niepożądane skutki. Na przykład w przypadku MECP2 korzystne może być wywołanie umiarkowanej aktywacji za pomocą tradycyjnego dCas9-VP64 zamiast silniejszych aktywatorów, ponieważ nadmierna aktywacja MECP2 może potencjalnie skutkować MDS. Opisany system CRISPRa został ostatnio zastosowany w kilku zwierzęcych modelach NDD.
Zespół Draveta jest chorobą NDD spowodowaną haploinsufficiencją kanału Na2 + bramkowanego napięciem SCN1A [ 72 , 73 ]. Poprzez celowanie w długi niekodujący RNA [ 74 ] lub region promotora genu SCN1A za pomocą CRISPRa [ 75 ], w badaniach pomyślnie zwiększono ekspresję SCN1A i przywrócono dysfunkcyjną pobudliwość neuronów i fenotypy napadów. Podobnie, haploinsufficiency kanału Na2 + bramkowanego napięciem SCN2A również powiązano z ASD [ 76,77 ] . W jednym z ostatnich badań wykorzystano CRISPRa do skutecznego przywrócenia pobudliwości neuronalnej i deficytów elektrofizjologicznych u myszy Scn2a +/- do poziomów typu dzikiego, przy czym efekt utrzymywał się odpowiednio przez co najmniej 3 i 8 miesięcy [ 78 ].
Poza kontekstem NDD, CRISPRa stosowano do celowania w kanał potasowy bramkowany napięciem KCNA1 w celu zapobiegania częstości napadów i dysfunkcji poznawczych w mysim modelu padaczki [ 79 ]. Na poziomie fenotypu fizjologicznego CRISPRa zastosowano również do zwiększenia transkrypcji genów ryzyka otyłości SIM1 i MC4R oraz do skutecznego ratowania haploinsufitowej otyłości w modelach mysich – a efekty utrzymywały się do 9 miesięcy po leczeniu [ 80 ]. Na poziomie ekspresji inne badanie wykazało, że CRISPRa skutecznie zwiększał ekspresję TTR u myszy transgenicznych dCas9-SAM 19 dni po leczeniu, ale efekt ten zmniejszył się w ciągu 8 miesięcy [ 81 ]. Z wyjątkiem tych badań, nadal nie ma wystarczających informacji na temat stopnia trwałości efektów terapii CRISPRa. Pomyślne tłumaczenie kliniczne wymaga dalszych badań in vivo, które oceniają znacznie późniejsze punkty czasowe.
Ratunek na poziomie mRNA
Ukierunkowane hamowanie hamujących NAT lub aktywacja genu przy użyciu CRISPRa to przykłady strategii terapeutycznych dla NDD na poziomie DNA. Jednakże możliwa jest również regulacja ekspresji NAT na poziomie potranskrypcyjnym. Badając kolejny poziom regulacji, warto ocenić potencjał wykorzystania ASO, gdyż mogą one zwiększać ekspresję genów poprzez różne mechanizmy. Mechanizmy te można podzielić na dwie główne kategorie funkcjonalne: (a) zwiększenie poziomu genu sensownego poprzez bezpośrednie interakcje z transkryptem mRNA genu sensownego oraz (b) zwiększenie poziomu genu sensownego poprzez hamowanie NAT za pośrednictwem ASO.
Bezpośrednia regulacja w górę genu sensownego
Badając zdolność ASO do zwiększania poziomu genu sensownego poprzez bezpośrednią interakcję z mRNA genu sensownego, pierwszy mechanizm leży w ASO, które celują w otwarte ramki odczytu (uORF) transkryptu sensownego [ 82 ] ( ryc. 3 ) . uORF to region w nieulegającym translacji regionie 5′ (UTR) transkryptu mRNA, który często zawiera dodatkowy kodon start, jak również kodon stop [ 83 ]. Kiedy translacja zostanie zainicjowana w tym loci, może nastąpić spadek efektywności translacji białka w miejscu startu powyżej z powodu wytwarzania peptydu, który ostatecznie blokuje funkcję rybosomów [ 84,85 ] . Aby wykorzystać ten mechanizm regulacji genu, zaprojektowano ASO tak, aby specyficznie celowały w uORF genu Lrpprc w modelach mysich i odkryły, że leczenie skutecznie zwiększyło ekspresję białka LRPPRC, co dostarczyło dowodów na skuteczność tego mechanizmu [ 84 ] Jednakże potrzebne są dalsze badania, aby zbadać skuteczność celowania w uORF w modelach chorób, ponieważ wiele z tych badań raczej potwierdziło zasady niż wykazało sukces w modelach chorób.

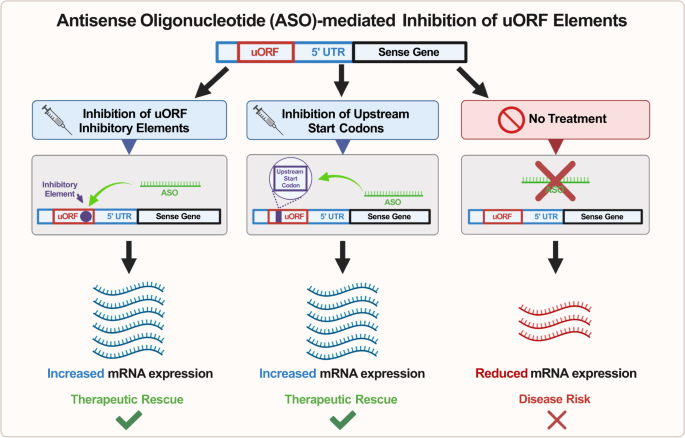
ASO można zastosować do hamowania kodonu start powyżej w otwartej ramce odczytu (uORF) transkryptu mRNA w celu zwiększenia ekspresji translacyjnej genu docelowego; B ASO można stosować do celowania i hybrydyzacji z elementami hamującymi, które tworzą struktury drugorzędowe hamujące translację w obrębie uORF transkryptu mRNA, aby ostatecznie zwiększyć ekspresję translacyjną genu docelowego. Zmniejszenie ekspresji mRNA w skrajnej prawej kolumnie jest wynikiem nieleczonego genu haploinsuficient.
Po drugie, zamiast pośredniczyć w cięciu transkryptów mRNA, ASO można również zaprojektować tak, aby celowały i hybrydyzowały z elementami hamującymi w regionie 5’UTR transkryptu mRNA. Wykazano wcześniej obecność struktur drugorzędowych hamujących translację w obrębie 5′ UTR transkryptów mRNA [ 82 , 86 ], a zaprojektowanie ASO komplementarnych do tych regionów mogłoby potencjalnie złagodzić hamowanie translacji (ryc. 3 ).
Na przykład w jednym badaniu wykorzystano ASO do nakierowania na strukturę typu spinki do włosów w regionie 5′ UTR mRNA LDLR , która hamowała translację białka, co skutkowało wzrostem ekspresji białka LDLR i wychwytu LDL w komórkach HEK293T [ 82 ]. Podobne metody zastosowano w przypadku mukowiscydozy (CF), choroby charakteryzującej się znacznymi dysfunkcjami płuc i trzustki, będącej wynikiem mutacji w genie CFTR kodującym kanał Cl [ 87 ] . W komórkowym modelu CF zaprojektowano ASO tak, aby celowały w hamujące drugorzędowe struktury mRNA w uORF 5’UTR transkryptu mRNA CFTR – skutecznie zwiększając zarówno ekspresję, jak i funkcję CFTR [ 88 ]. Jednakże, chociaż sukces w modelu CF in vitro zapewnia potencjał do przełożenia klinicznego, potrzebne są dalsze dowody na sukces in vivo przy zastosowaniu tego specyficznego podejścia terapeutycznego.
Po trzecie, istnieją dowody na skuteczne przywrócenie nieprawidłowego splicingu mRNA w stanach mięśni niezwiązanych z NDD, takich jak rdzeniowy zanik mięśni (SMA) [ 89 ] i dystrofia mięśniowa Duchenne’a (DMD) [ 90 ] poprzez celowanie w połączenia splicingowe i elementy cis-regulacyjne za pomocą ASO [ 91 , 92 ] (ryc. 4A ). W SMA u poszczególnych osób brakuje działającej kopii genu SMN1 , dlatego ASO wykorzystuje się do ułatwienia prawidłowego splicingu genu SMN2 poprzez indukcję włączenia eksonu 7, co ostatecznie ratuje ekspresję białka SMN w modelach komórek ssaczych in vitro [ 91] . ]
Ryc. 4: Schemat mechanizmów regulacji alternatywnego splicingu przez oligonukleotydy antysensowne (ASO).

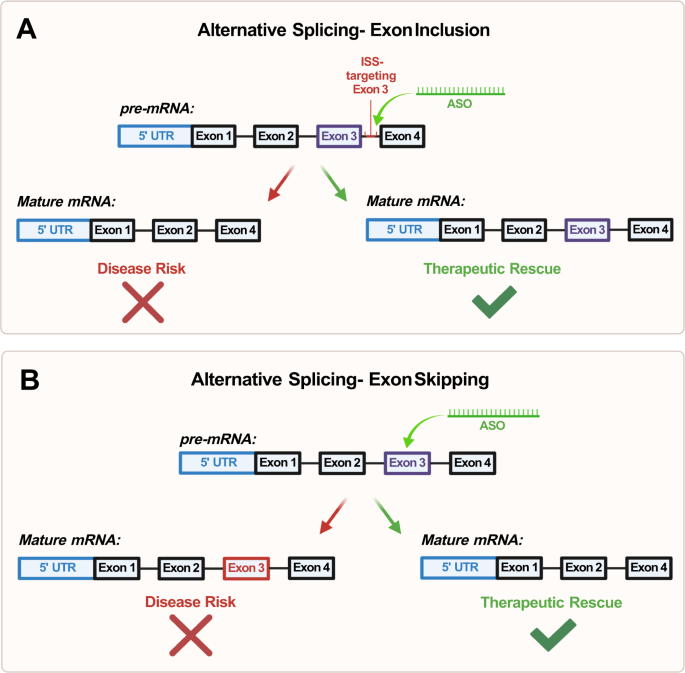
Inkluzja egzonu działa poprzez ukierunkowanie na połączenia splicingowe i elementy regulatorowe cis (CRE) jako forma alternatywnego splicingu transkryptu mRNA, tworząc w ten sposób funkcjonalną kopię białka. W tym przykładzie ASO celuje w tłumik intronowego splicingu (ISS); Pomijanie eksonu B jest formą regulacji alternatywnego splicingu, która polega na maskowaniu dysfunkcjonalnego eksonu za pomocą ASO, co skutkuje wycięciem eksonu z końcowego transkryptu mRNA. Pozwala to na przywrócenie ramki odczytu i translację częściowo funkcjonalnego białka.
W 2016 roku FDA zezwoliła na stosowanie Spinrazy, pierwszej terapii opartej na leku w leczeniu SMA [ 93 ]. Spinraza działa dzięki mechanizmowi ukierunkowanego włączenia eksonu 7 do mRNA SMN2, skutecznie ratując funkcje motoryczne u pacjentów [ 94 , 95 ]. W DMD mutacje w genie dystrofiny powodują przesunięcie ramki odczytu i prowadzą do wytworzenia niefunkcjonalnego białka dystrofiny [ 96 ]. ASO ukierunkowane na DMD są ukierunkowane na eksony niosące mutacje przesunięcia ramki odczytu odpowiedzialne za DMD. Zaprojektowane ASO maskują dysfunkcjonalny ekson w pre-mRNA, w wyniku czego ekson zostaje odłączony od końcowego transkryptu mRNA, przywracając w ten sposób ramkę odczytu i wytwarzając częściowo funkcjonalną kopię białka dystrofiny [ 92 , 97 ] (ryc. 4B ).
Chociaż we wczesnych stadiach badań klinicznych z zastosowaniem ASO Drisapersenu pomijającego egzony w leczeniu DMD pokładano wiele nadziei, badania fazy 3 nie przyniosły sukcesu klinicznego [ 98 , 99 , 100 ]. Jednak w 2016 r. FDA zezwoliła na stosowanie Eteplirsenu, ASO wywołującego pomijanie eksonów w celu ekspresji częściowo funkcjonującej dystrofiny, jako pierwszej terapii lekowej DMD [ 101 ]. Podobnie ASO promujący alternatywny splicing o nazwie Milasen został zaprojektowany jako spersonalizowany lek do leczenia osób cierpiących na chorobę Battena, chorobę neurodegeneracyjną charakteryzującą się ślepotą, zwiększoną podatnością na drgawki i opóźnieniem rozwoju [ 102 , 103 , 104 ]. Mutacja w genie MFSD8 (znanym również jako CLN7 ) spowodowała powstanie skróconego i dysfunkcyjnego białka, a leczenie Milasenem było w stanie skutecznie pomóc w leczeniu fenotypów napadów padaczkowych i poprawić wyniki neurologiczne, tymczasowo poprawiając jakość życia pacjenta [ 104 ].
Po czwarte, ASO mogą tłumić rozpad transkryptów mRNA za pośrednictwem nonsensu (NMD) poprzez celowanie w region kompleksu połączenia egzonu (EJC) zlokalizowany poniżej kodonów przedwczesnej terminacji transkryptu (ryc. 5A ). Mechanicznie NMD zależy od obecności co najmniej jednego EJC, a ukierunkowanie na ten szlak za pomocą ASO doprowadziło do wzrostu ekspresji genu MECP2 w modelach komórek ssaczych in vitro [ 105 ]. Dostarcza to dowodów na potencjalne wykorzystanie ASO do hamowania NMD MECP2 , otwierając w ten sposób drogę terapeutyczną w leczeniu zespołu Retta. Jednakże ponownie ważne jest, aby zwrócić uwagę na wrażliwość dawkowania MECP2 jako istotną przeszkodę na drodze do skutecznego leczenia. Ponadto potrzebne są dalsze badania w celu określenia idealnej dawki zapobiegającej indukcji MDS, a przed pomyślnym przełożeniem klinicznym na modele in vivo potrzebne są dalsze badania.
Ryc. 5: Schemat mechanizmów oligonukleotydów antysensownych (ASO) związanych z degradacją transkryptu mRNA.

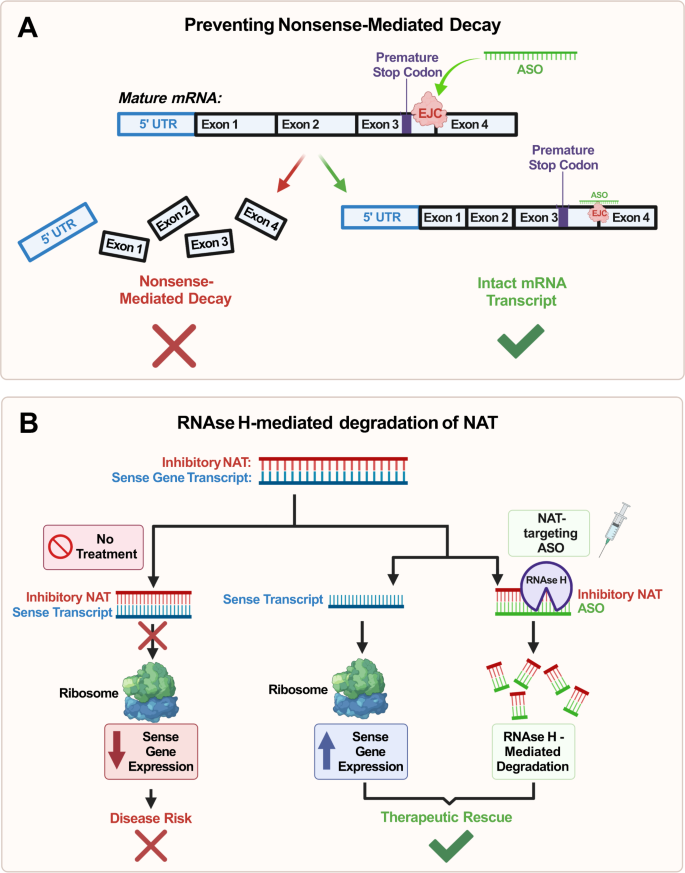
ASO zapobiegają rozpadowi za pośrednictwem nonsensu (NMD) transkryptu mRNA poprzez hybrydyzację. Celując w region kompleksu połączenia egzonu (EJC) poniżej kodonów przedwczesnej terminacji transkryptu (PTC), ASO zwiększają stabilność transkryptu. Zapobiega to NMD i zwiększa ekspresję translacyjną genu docelowego; B ASO mogą indukować za pośrednictwem RNAzy H degradację naturalnego transkryptu antysensownego (NAT), aby złagodzić hamowanie sensownego transkryptu mRNA poprzez hybrydyzację. Enzym endonukleaza RNAza H rozpozna i rozszczepi dupleks ASO-NAT, umożliwiając w ten sposób niehamowaną translację odpowiedniego genu sensownego. Aktywacja allelu ojcowskiego UBE3A poprzez ASO ukierunkowane na UBE3A-AS (najbardziej prawa kolumna) jest konkretnym przykładem tego mechanizmu stosowanego w leczeniu zespołu Angelmana.
Regulacja w górę genu sensownego poprzez degradację NAT
Chociaż wspomniane wcześniej mechanizmy działają w celu zwiększenia ekspresji genu sensownego poprzez bezpośrednie oddziaływanie na transkrypt mRNA genu sensownego, jednym ograniczeniem jest fakt, że uORF występują jedynie w około 50% mRNA ssaków i w obrębie tych 50% nie wszystkie mają działanie hamujące [ 106 , 107 ]. Dlatego też regulacja w górę genów sensownych związanych z ASD poprzez degradację NAT za pośrednictwem ASO ma dużą wartość. Mechanicznie, gdy ASO zwiąże się z transkryptem mRNA NAT, enzym endonukleaza RNAzy H rozszczepi dupleks RNA, skutecznie tłumiąc funkcję hamującą tych NAT [ 59 , 108 ] (ryc. 5B ).
Jak omówiono wcześniej, zespół Draveta (DS) jest chorobą NDD spowodowaną haploinsufficiencją kanału Na2 + bramkowanego napięciem SCN1A [ 72 , 109 ]. Dzięki zastosowaniu ASO do ukierunkowania na SCN1A NAT w jednym badaniu udało się skutecznie uratować sensowną ekspresję genu SCN1A , złagodzić pobudliwość neuronów i drgawki w mysim modelu DS – efekt, który prawie całkowicie odzwierciedla uratowane fenotypy obserwowane podczas leczenia CRISPRa [ 74 , 75 ].
Podobnie zespół Angelmana (AS) jest chorobą NDD wynikającą z haploinsuficiencji genu UBE3A . Chociaż poprzednie sukcesy odkryto w ratowaniu opartym na CRISPR na poziomie DNA, możliwe jest również namierzenie transkryptu mRNA UBE3A NAT w celu degradacji na poziomie transkrypcji [ 60 ]. Liczne badania wykazały , że możliwe było zmniejszenie ekspresji UBE3A NAT, uratowanie sensownej ekspresji UBE3A oraz polepszenie fenotypów poznawczych i behawioralnych w mysim modelu AS poprzez degradację za pośrednictwem RNAzy [ 59,110 ]. Na poparcie tego mechanizmu eksperymentalne leki GTX-102 ( https://clinicaltrials.gov/ct2/show/NCT04259281 ), ION582 ( https://clinicaltrials.gov/ct2/show/NCT05127226 ) i RO7248824 ( https: //clinicaltrials.gov/ct2/show/NCT04428281 ) wykorzystują ASO ukierunkowane na NAT i obecnie przechodzą badania kliniczne fazy 1/2 z udziałem dzieci i dorosłych pacjentów z AS. Należy jednak zauważyć, że badanie GTX-102 zostało tymczasowo wstrzymane ze względu na rozwój u pacjentów ostrej poliradikulopatii zapalnej w wyniku leczenia [ 111 ]. Donoszono, że poliradikulopatia nie była reakcją immunologiczną, ale prawdopodobnie wynikiem miejscowej toksyczności spowodowanej wysokim stężeniem GTX-102 w miejscu wstrzyknięcia [ 111 ]. Pomimo tych działań niepożądanych lek okazał się bardzo obiecujący, ponieważ wszyscy uczestnicy wykazali znaczną poprawę w zakresie funkcji motorycznych, komunikacji, snu i problemów behawioralnych.
Ukierunkowane terapie edytujące RNA
Poza terapeutykami opartymi na ASO, które działają głównie jako modulatory potranskrypcyjne, przyszłość może wiązać się z bezpośrednią edycją transkryptu mRNA. Przy pomocy Cas13, rybonukleazy kierowanej przez RNA, tradycyjnie stosowanej do skutecznego i specyficznego cięcia transkryptów mRNA, możliwe było opracowanie nowego białka fuzyjnego zdolnego do edycji transkryptu mRNA, znanego jako edycja RNA w celu programowalnej zamiany A na I (NAPRAWA). [ 112 ] Aby opracować REPAIR, ortolog Cas13b zmutowano tak, aby był katalitycznie martwym Cas13b (dCas13b) i połączono z deaminazą adenozynową działającą na enzym RNA 2 (ADAR2), co umożliwiło zastąpienie reszt adenozyny (A) resztami inozyny (I) w mRNA transkrypcje. Podobnie udowodniono, że fuzja enzymu dCas9 z ADAR2 powoduje te same modyfikacje A do I w mRNA z porównywalną wydajnością i specyficznością jak system REPAIR [ 113 ]. Opierając się na edycji RNA związanej z ADAR2, w jednym z ostatnich badań opracowano AAV, które koeksprymują ludzką domenę katalityczną ADAR2 typu dzikiego połączoną z peptydem λN bakteriofaga (Editase wt ) i RNA kierującym Mecp2 [ 114 ]. W badaniu wykorzystano system oparty na Editase wt do namierzenia mutacji Mecp2 G311A , która tworzy kodon stop, co skutkuje brakiem białka MeCP2. Wstrzyknięcie konstruktu pozaoczodołowego myszom z mutacją Mecp2 G311A skutecznie przywróciło ekspresję i funkcję białka MeCP2, wydłużyło przeżycie i poprawiło funkcję oddechową myszy. Chociaż te oparte na ADAR środki terapeutyczne do edycji RNA są wysoce specyficzne w dokonywaniu modyfikacji A do I w transkryptach mRNA, nadal zapewniają obiecującą alternatywną drogę terapeutyczną do klinicznej translacji środków terapeutycznych NDD.
Ratunek na poziomie białka za pomocą leków małocząsteczkowych
W dalszej części głównego dogmatu biologii kluczowe podejście terapeutyczne polega na aktywacji lub hamowaniu szlaków molekularnych związanych z NDD. Skutecznie przesunęłoby to uwagę z terapii opartych na ekspresji na terapie potranslacyjne.
Szlaki proliferacyjne
Stwardnienie guzowate (TS) to choroba monogenowa charakteryzująca się dużą częstością występowania ASD, spowodowaną mutacjami LoF w genach TSC1 i TSC2 [ 115 ]. Mutacje te prowadzą do nadmiernej aktywacji szlaku mTOR, co ostatecznie prowadzi do powstania łagodnych nowotworów w wielu narządach [ 116,117 ] . Badania na myszach wykazały, że inhibitory mTOR, takie jak rapamycyna, która wcześniej uzyskała zgodę FDA na leczenie raka, skutecznie eliminowały braki w zachowaniach społecznych i powtarzalnych u myszy z mutacją TSC w 7. dniu po urodzeniu i jedynie deficyty społeczne u 6-tygodniowych mutantów TSC myszy [ 118 , 119 ]. Jednakże translacyjne próby hamowania mTOR nie powiodły się w zakresie poprawy funkcjonowania poznawczego, problemów behawioralnych, autyzmu i deficytów neuropsychologicznych u dzieci w wieku 4-17 lat leczonych ewerolimusem – innym inhibitorem mTOR, który został wcześniej zatwierdzony przez FDA do leczenia raka [ 120 ]. Biorąc pod uwagę te wyniki, ważne jest, aby wziąć pod uwagę moment podania leku, ponieważ wcześniejsza interwencja może zapewnić większą skuteczność leczenia, o czym świadczy zależny od wieku sukces w badaniach na myszach.
Ratowanie hamujących szlaków sygnałowych
Wcześniej postawiono hipotezę, że potencjalną utratę równowagi neuronalnych sygnałów pobudzających/hamujących [ 121 , 122 , 123 ] w obrębie podtypów syndromicznych ASD, takich jak FXS, RTT, AS i w idiopatycznym ASD, można przypisać zmniejszeniu hamującego GABA A funkcja receptora [ 124 , 125 ]. Zatem zwiększenie lub wzmocnienie funkcji receptora GABAA może być potencjalnym celem terapeutycznym w ratowaniu fenotypów ASD. Niedawno pojawił się arbaklofen, agonista GABA B , jako potencjalny lek na ASD. W mysim modelu z delecją 16p11.2 stwierdzono, że arbaklofen pomaga w leczeniu deficytów pamięci, mierzonych na podstawie zachowania polegającego na zawieszaniu się w zależnych od kontekstu zadaniach uczenia się awersyjnego [ 126 ]. To samo badanie wykazało, że arbaklofen skutecznie łagodził deficyty w interakcjach społecznych między mężczyznami i kobietami, mierzone na podstawie wąchania od nosa do odbytu i odbytu oraz czasu, jaki mężczyzna spędzał w podążaniu za samicą w okresie rui. W małym badaniu klinicznym z udziałem 25 nastolatków stwierdzono, że arbaklofen ratuje powolne przetwarzanie sensoryczne słuchowe u mężczyzn z idiopatycznym ASD [ 127 ]. Należy jednak wziąć pod uwagę, że chociaż idiopatyczne badanie ASD odniosło pewien sukces, badania kliniczne III fazy z udziałem dzieci (w wieku 5–11 lat) i młodzieży do dorosłych (w wieku 12–50 lat) pacjentów z FXS zakończyły się niepowodzeniem, ponieważ nie byli w stanie osiągnąć podstawowego rezultatu, jakim było ratowanie deficytów społecznych [ 128 ]. Wyniki tego badania klinicznego fazy III sugerują, że przyszłe badania powinny uwzględniać wyższe dawki, większą liczebność próby, młodszą grupę wiekową i lepsze mierniki wyników. Dlatego też, chociaż stymulacja receptora GABAB przy użyciu arbaklofenu w celu zwiększenia hamujących sygnałów neuronalnych i ratowania nieprawidłowej równowagi pobudzenia/hamowania w ASD może okazać się obiecująca, heterogeniczność genetyczna ASD może utrudniać jego dalsze stosowanie. W przyszłych badaniach klinicznych dotyczących ASD wyraźnie potrzebna jest stratyfikacja genetyczna pacjentów i lepsze pomiary wyników klinicznych.
Ratowanie pobudzających szlaków sygnałowych
Alternatywna hipoteza głosi, że wzrost pobudzającej sygnalizacji glutaminianu może odgrywać rolę w rozregulowaniu równowagi neuronalnych sygnałów pobudzających/hamujących. W FXS stwierdzono, że nastąpił przyczynowy wzrost metabotropowego receptora glutaminianu 5 (mGluR5), który towarzyszy utracie ekspresji FMR1 [ 129 ]. Hamowanie molekularne w loci mGluR5 było skuteczne w wielu modelach mysich. U myszy BTBR zastosowanie antagonisty mGluR5, MPEP, skutecznie uratowało fenotyp powtarzalnej pielęgnacji [ 130 ]. MPEP uratował także nie tylko tę samą powtarzalną pielęgnację, ale także związane z lękiem zakopywanie marmurów i poruszanie się w mysim modelu kwasu walproinowego (VPA) [ 131 ]. Co więcej, leczenie z użyciem negatywnego modulatora allosterycznego (NAM) mGluR5 CTEP uratowało upośledzoną pamięć w mysim modelu mikrodelecji 16p11.2 [ 132 ]. Tłumaczenie kliniczne okazało się jednak wyzwaniem, a badania przedkliniczne nad NAM mGluR5 nie pozwoliły na uratowanie fenotypów u ludzkich pacjentów z FXS [ 133 ]. Niepowodzenia w badaniach przedklinicznych można prawdopodobnie przypisać niewłaściwej ekstrapolacji dawek z modeli mysich, zbyt krótkiemu czasowi leczenia lub temu, że hamowanie mGluR5 jest preferencyjnie skuteczne w młodszej populacji – parametry te można dostosować w przyszłych badaniach [ 134,135 ] Wczesne, ciągłe hamowanie sygnalizacji mGlu grupy 1 częściowo uratowało nieprawidłowości kręgosłupa dendrytycznego w mysim modelu zespołu łamliwego chromosomu X z nokautem Fmr1 [ 133 ].
Inna niedawno odkryta strategia terapeutyczna w leczeniu ASD polega na celowaniu w małą GTPazę, RhoA, która bierze udział w strukturze i ruchliwości cytoszkieletu komórkowego [ 136 ] i stwierdzono, że jej poziom jest podwyższony w niektórych modelach ASD, podczas gdy w innych jest obniżony [ 34,137,138 , 139 ]. Cullin3 ( Cul3 ) jest genem istotnym dla całego genomu ryzyka ASD [ 26 ], którego haploinsuficiency przyczynia się do zmniejszonego wzrostu dendrytów neuronalnych i zmniejszonej aktywności sieci neuronowej [ 34 ]. Ponieważ mysi model z haploinsufitem Cul3 również wykazał zwiększoną ekspresję RhoA, zastosowano leczenie z użyciem inhibitora RhoA Rhosin, aby skutecznie uratować te fenotypy in vitro w hodowlach pierwotnych neuronów korowych pochodzących od tych myszy [ 34 ]. W mysim modelu stresu afektywnego, opartym na porażce społecznej, stwierdzono również, że Rhosin ratuje fenotypy behawioralne, takie jak upośledzone nowe interakcje myszy i zachowania unikające poprzez supresję receptora dopaminy 1 średnich neuronów kolczastych (D1-MSN) w jądrze półleżącym ( NAc) [ 140 ]. Podobnie u myszy Kctd13 (gen w 16p11.2 CNV) z haploinsuficientem i nokautem nastąpił wzrost ekspresji RhoA w połączeniu z niedoborami sygnalizacji synaptycznej – efekt, który został złagodzony po leczeniu Rhosin [ 137 ]. Te sukcesy w modelach mysich dostarczają dowodów, że Rhosin może być ważną metodą terapeutyczną dla osób z delecją w loci 16p11.2 lub Cul3 .
Dostawa środków leczniczych
Wirusowe dostarczanie konstruktów genowych CRISPR-Cas9 i ASO jest powszechnie stosowaną metodą wykorzystującą wektory, takie jak lentiwirusy, adenowirusy i wirusy związane z adenowirusami (AAV) [ 141 ] (Tabela 1 ). Chociaż są one powszechnie stosowane, ważne jest, aby wziąć pod uwagę potencjalne wady, takie jak odpowiedź immunologiczna danej osoby na te wektory, a także ograniczenia dotyczące wielkości opakowań. Chociaż wektor adenowirusowy może mieć większy limit upakowania do ~36 kb, istnieje większe ryzyko zapalnej odpowiedzi immunologicznej [ 142 ]. Z drugiej strony AAV mają ograniczony rozmiar opakowania wynoszący ~5 kb i charakteryzują się znacznie łagodniejszym ryzykiem zapalenia [ 143 ]. Innym ważnym wektorem wirusowym jest lentiwirus, który jest rodzajem retrowirusa o rozmiarze opakowania wynoszącym ~9 kb – co stanowi wartość pośrednią pomiędzy wektorami adenowirusa i AAV [ 144 ]. Jedną z głównych zalet lentiwirusa jest jego zdolność do dostarczania transgenów i integrowania ich z genomem w celu długotrwałej ekspresji. Jednakże ta zaleta zwiększa ryzyko wystąpienia efektów ubocznych w wyniku mutagenezy insercyjnej, ponieważ lentiwirusy nie mają wysokiej specyficzności. Mutageneza insercyjna jest wynikiem integracji egzogennego DNA lentiwirusa z genomem gospodarza w otwartych regionach, a jeśli nastąpi to w miejscu innym niż docelowy, może skutkować nieprawidłową ekspresją genów [ 145 ]. Badania wykazały również, że wektor lentiwirusowy stwarza umiarkowane ryzyko zapalenia, ale potrzebne są dalsze badania nad immunogennością i zapobieganiem zdarzeniom rekombinacyjnym w celu optymalizacji translacji klinicznej [ 146 ].
Pomimo wcześniejszych przekonań, że AAV nie podlegają mutagenezie insercyjnej, niektóre badania sugerują, że AAV mają tendencję do przypadkowej integracji z genomem podczas pęknięć podwójnej nici (DSB) [ 147 ], co prowadzi do powstawania nowotworów i rozrostu komórek wątroby u zwierząt [ 148 ]. Na przykład w mysim modelu mukopolisacharydozy (MPS VII) dostarczenie noworodkom myszy transgenu b-glukuronidazy przy użyciu wektora AAV2 spowodowało, że u myszy rozwinął się rak wątrobowokomórkowy (HCC) – z dowodem integracji AAV z guzami [ 149,150 ] Większość integracji AAV wystąpiła w RNA odciśniętym i zgromadzonym w jądrze ( locus Rian) , co przyczynia się do przejścia nabłonka do mezenchymalnego w przypadku raka [ 148 ]. Badania te sugerują również, że wiek odgrywa rolę w rozwoju HCC indukowanym przez AAV, ponieważ Rian wyraża się w większej ilości na początku życia. W modelu psa chorego na hemofilię stwierdzono integrację AAV cztery lata po leczeniu psim czynnikiem VIII (cFVIII) w wektorze AAV8/9, jednakże nie było dowodów na powstawanie nowotworu u tych psów, które miały integrację AAV . .
Integracyjna skłonność AAV jest szczególnie ważna do rozważenia w świetle terapii polegających na edycji genomu. Ponieważ stwierdzono, że AAV integrują się z DSB z dużą częstotliwością [ 152 ] , przed translacją kliniczną należy dokładnie ocenić połączenie CRISPR-Cas9 z AAV jako wektorem dostarczającym [ 58,153 ]. Chociaż dotychczas nie zgłoszono żadnych potwierdzonych zdarzeń genotoksycznych u ludzi w wyniku stosowania wektorów rAAV, biorąc pod uwagę powyższe dowody, konieczne będzie przeprowadzenie dalszych badań na większych organizmach modelowych (takich jak NHP) w dłuższych punktach czasowych. Oprócz tego konieczne będzie ciągłe monitorowanie i testowanie biomarkerów HCC w warunkach klinicznych.
Wiele wektorów wirusowych nie przenika przez BBB z dużą skutecznością i należy je wprowadzić poprzez inwazyjne bezpośrednie wstrzyknięcie lub potencjalnie neurotoksyczne rozerwanie BBB [ 154 ]. Ponieważ większość genów ryzyka NDD ulega ekspresji w mózgu, suboptymalne metody dostarczania tych wektorów wirusowych mogą wydawać się nieodpowiednie do klinicznego dostarczania leków NDD. Chociaż niektóre serotypy AAV, takie jak AAV1, AAV2, AAV5 i AAV8, mogą transdukować neurony, nie są one w stanie skutecznie przenikać przez BBB poprzez nieinwazyjne wstrzyknięcie dożylne [ 155 ]. Stwierdzono jednak, że serotyp AAV9 ma zdolność skutecznego przenikania przez BBB po podaniu dożylnym noworodkom i dorosłym myszom [ 156 ]. Dlatego też, w przeciwieństwie do adenowirusa, lentiwirusa i innych serotypów AAV, wektor AAV9 jest w stanie skutecznie uniknąć dwóch głównych problemów: konieczności inwazyjnego wstrzyknięcia i naruszenia integralności BBB, chociaż jego skuteczność transdukcji maleje wraz z wiekiem [ 156 ] . W modelu mysim RTT stwierdzono, że wewnątrzczaszkowe dostarczanie transgenu MECP2 za pośrednictwem AAV9 skutecznie zwiększa przeżycie i niektóre fenotypy behawioralne [ 43 ]. W badaniu klinicznym III fazy wektor AAV9 odniósł sukces w dożylnym dostarczaniu transgenu SMA1 , skutecznie przywracając funkcje motoryczne u pacjentów z SMA [ 157 , 158 ]. Konieczne są dalsze badania, aby szczegółowo zbadać różnice w skuteczności transdukcji środków terapeutycznych pomiędzy zastrzykami dożylnymi i doczaszkowymi/dooponowymi w organizmach modelowych.
Chociaż tradycyjnie głównym wektorem używanym do dostarczania konstruktów CRISPR-Cas9 były AAV, stosunkowo mały rozmiar opakowania (~4,5 kb) sprawia, że badacze muszą oddzielnie dostarczać dwa oddzielne wektory zawierające konstrukt CRISPR-Cas9 (~4,2 kb) i gRNA . W szczególności w podejściu CRISPRa aktywatory transkrypcji, które są połączone z enzymem dCas9, zwiększą długość konstruktu, tak że kombinacja sgRNA i aktywatorów transkrypcji dCas9 będzie zbyt duża, aby można ją było załadować na pojedynczy wektor AAV. Jest to mniej pożądane w warunkach klinicznych, ponieważ wymagałoby wytwarzania dwóch AAV, co znacznie zwiększa koszty opracowania leku.
Biorąc pod uwagę, że tradycyjny system CRISPR-Cas9 wywodzi się z bakterii Streptococcus pyogenes (SpCas9), najnowsze techniki miały na celu przezwyciężenie tego ograniczenia wielkości poprzez badanie skuteczności innych mniejszych ortologów Cas9. Jeden z takich przykładów obejmuje wykorzystanie mniejszego ortologa Cas9 ze Staphylococcus aureus (SaCas) ( 3,15 kb), który jest o ~1 kb krótszy niż SpCas9, tak że konstrukt CRISPR-Cas9 i gRNA mogą zmieścić się w pojedynczym wektorze AAV [ 159,160 ] Technika ta skutecznie przywróciła ekspresję sensownego genu UBE3A poprzez degradację NAT w mysim modelu AS, jak również uratowała fenotypy choroby w mysim modelu DMD [ 58,161 ] . Podobnie Cas9 z Campylobacter jejuni (CjCas9) (2,95 kb) zastosowano w wektorach z pojedynczym AAV, aby skutecznie zmniejszyć fenotyp neowaskularyzacji naczyniówkowej w mysim modelu zwyrodnienia plamki związanego z wiekiem (AMD) [ 162 ]. Dalsze eksperymenty powinny zbadać skuteczność CjCas9 w ratowaniu fenotypów związanych z NDD.
Ponieważ BBB zapobiega również przedostawaniu się do OUN większości leków małocząsteczkowych, oprócz wspomnianych wcześniej wektorów wirusowych, niedawny rozwój wariantów AAV9 okazał się potencjalnym kluczem do rozwiązania tego problemu. Warianty takie jak rAAV-PHP.B i rAAV-PHP.eB drugiej generacji zawierają zmodyfikowany kapsyd, który przyczynia się do niespotykanej dotąd skuteczności przekraczania bariery krew-mózg po wstrzyknięciu dożylnym, wraz z dużą zdolnością dyfuzji zarówno do neuronów, jak i do komórek glejowych myszy [ 163 , 164 , 165 , 166 ]. Wysoka skuteczność krzyżowania przez BBB rAAV-PHP.B i rAAV-PHP.eB wynika z ich interakcji z białkiem LY6A (znanym również jako SCA-1) ulegającym ekspresji na komórkach śródbłonka mikronaczyniowego mózgu (BMVEC) mysiego BBB [ 167 ]. Niestety, LY6A nie ulega ekspresji u naczelnych, co stanowi główną przeszkodę translacyjną dla przenikania ludzkiego BBB. W świetle tych przeszkód translacyjnych dla rAAV-PHP.eB i rAAV-PHP.B, w jednym z ostatnich badań uzyskano AAV.CPP16 poprzez wstawienie peptydów penetrujących komórki (CCP) do kapsydu AAV9 pomiędzy aminokwasami Q588 i A589 [ 168 ]. W porównaniu z niezmodyfikowanym AAV9, dożylne wstrzyknięcie AAV.CPP16 myszom wykazało znacznie większą skuteczność krzyżowania przez BBB, większą wydajność transdukcji i wyższą specyficzność neuronów w obrębie wielu szczepów myszy i makaków cynomolgus. Chociaż wektor AAV.CPP16 jest obiecujący dla przyszłości terapii ukierunkowanych na OUN, potrzebne są dalsze badania, aby potwierdzić skuteczne ratowanie w modelach chorobowych.
Lipidoidy pochodzące z neuroprzekaźników (NT-lipidoidy) stały się ostatnio nowym obiecującym wektorem dostarczania, zwłaszcza do OUN. Jedno z badań wykazało niedawno, że wektor NT-lipidoidowy z powodzeniem wprowadził ukierunkowany na Tau ASO, lek małocząsteczkowy i białko fuzyjne do mózgu myszy poprzez wstrzyknięcie dożylne [ 169 ]. Podobnie nanocząsteczki można potencjalnie wykorzystać do dostarczania leków do mózgu w ramach dwuetapowej strategii celowania, która wykorzystuje wysoką nieprzepuszczalność BBB do selektywnego zatrzymywania znaczników ligandów na powierzchni śródbłonka mózgu [ 170 ]. Dowody z innych badań wykazały, że ASO, mRNA, konstrukty CRISPR-Cas9 i leki małocząsteczkowe można dostarczać za pomocą wektorów nanocząstek lipidowych [ 171 , 172 , 173 , 174 ]. Dlatego opracowanie lepszych wektorów do ukierunkowania specyficznego dla mózgu i neuronów mogłoby w dalszym ciągu udoskonalać strategie terapeutyczne NDD.
Dyskusja
W tym przeglądzie zbadano trzy główne cele interwencji w przypadku NDD w oparciu o główny dogmat, w szczególności na poziomach: DNA, mRNA i białka. Chociaż w modelach in vitro i zwierzęcych odniesiono duży sukces na poziomie DNA i białka, nie udało się jeszcze zastosować tych leków w badaniach klinicznych na ludziach. Różne techniki terapeutyczne omówione w tym przeglądzie, które były w trakcie badań klinicznych lub zakończyły się pomyślnie, podsumowano w Tabeli 2 .
Wysiłki mające na celu leczenie haploinsuficiencji genów będą wymagały dokładnego rozważenia specyficzności typu komórki, specyficzności tkanki i modulowania ekspresji prawidłowych izoform splicingowych – coś, co będzie się różnić w zależności od tkanki i etapów rozwojowych [ 175 ]. Alternatywne splicing odgrywa ważną rolę w NDD, a geny ryzyka ASD wykazują różną ekspresję izoform (DEI) na każdym etapie rozwoju prenatalnego [ 176 , 177 , 178 ]. Co więcej, DEI wpływają na ścieżki zaangażowane w rozwój dendrytów, organizację synaps i projekcję neuronalną, czyli procesy, które są rozregulowane w przypadku ASD [ 12 , 177 ]. Jedno z badań wykazało, że w neuronach pochodzących z pluripotencjalnych komórek macierzystych indukowanych przez człowieka NRXN1 +/- (hiPSC) nastąpił znaczny wzrost ekspresji nowych izoform NRXNα , w połączeniu ze spadkiem izoformy NRXNα typu dzikiego [ 179 ]. To rozregulowanie równowagi izoformy NRXNα spowodowało zmniejszenie aktywności neuronów i zakłócenia w dojrzewaniu neuronów. Dlatego też podczas procesu opracowywania terapii konieczne jest uwzględnienie wieku pacjenta, izoform specyficznych dla typu komórki, alternatywnych miejsc startu i alternatywnych promotorów genów docelowych. Siła promotora w terapii transgenowej lub modulacja aktywatorów transkrypcji CRISPRa będzie odgrywać kluczową rolę w określeniu odpowiedniego leczenia i uniknięciu nieprzewidywalnych lub niepożądanych konsekwencji leczenia.
Biorąc pod uwagę niedawne wysiłki polegające na skupieniu się na ratowaniu terapeutycznym genów w NDD, możliwe, że następna dekada przyniesie sukces translacyjny. Sukces ten zależy od dalszej optymalizacji technik będących jeszcze w powijakach, takich jak CRISPRa lub ASO oparte na uORF/hamujące NMD. Co więcej, mechanizmy terapeutyczne, takie jak ASO alternatywnego splicingu, które mają aprobatę FDA dla patologii innych niż NDD, takich jak Spinraza (SMA) i Eteplirsen (DMD), mogą stanowić solidną podstawę do dalszych badań z wykorzystaniem tych ASO dla genów z mutacjami splicingowymi związanymi z NDD. Jak dowodzą leki hamujące mTOR w leczeniu TSC, moment podania leku jest również ważnym czynnikiem wpływającym na pomyślne zastosowanie kliniczne.
W przypadku NDD niezwykle ważne będzie jak najszybsze zdiagnozowanie choroby, aby móc zastosować leki w krytycznych okresach rozwoju mózgu. Mając to na uwadze, badania wykazały, że okres od połowy płodu do wczesnego okresu poporodowego jest krytycznym oknem dla rozwoju neurologicznego, w którym ulega ekspresji wiele genów ryzyka ASD [ 180 , 181 , 182 ]. Przykład takiego można zaobserwować w badaniu, w którym zależna od Cre aktywacja Ube3a u embrionalnych myszy Ube3a Stop/p+ przywróciła wszystkie związane z AS deficyty motoryczne, behawioralne i neurologiczne, podczas gdy reaktywacja Ube3a u myszy po urodzeniu wykazała malejącą skuteczność wraz z wiekiem w zakresie motoryki. koordynacja ratownictwa [ 183 ] . Wraz z rozwojem strategii podawania leków ukierunkowanych na OUN i potencjalnymi powikłaniami związanymi z dostarczaniem leków w okresie prenatalnym, NDD będzie prawdopodobnie najskuteczniejsze w leczeniu we wczesnym okresie poporodowym. Należy zauważyć, że te punkty czasowe nie będą miały uniwersalnego zastosowania do wszystkich genów i wszystkich fenotypów. Inne badania wykazały skuteczne wyleczenie deficytów neurologicznych i elektrofizjologicznych u dorosłych myszy Ube3a Stop /p+ po reaktywacji genu Ube3a oraz uratowanie defektów neurologicznych u dorosłych myszy Mecp2 lox-Stop/y po reaktywacji genu Mecp2 [ 183,184,185 ] Podobnie, wcześniej omawiane badanie Scn2a CRISPRa skutecznie uratowało deficyty elektrofizjologiczne u dorastających myszy z haploinsufitem Scn2a [ 78 ]. Ta heterogeniczność ratowania fenotypowego w różnych punktach czasowych rozwoju sugeruje, że okresy interwencji terapeutycznej będą musiały być oceniane w oparciu o gen po genie.
Ponieważ modele mysie nie są w stanie dokładnie odzwierciedlić okresów neurorozwoju człowieka, kolejnym krokiem translacyjnym mogłoby być opracowanie modeli haploinsufficiencji w obrębie NHP – czegoś, co zostało już ustalone dla genów SHANK3 i MECP2 u makaków cynomolgus [ 35 , 36 ]. Marmozeta zwyczajna ( Callithrix jacchus ) to kolejny potencjalny organizm modelowy ludzkiej NDD, a badania wykazały, że ekspresja genów i wzorce dystrybucji genów w mózgach ludzi i marmozet były bardziej podobne niż u ludzi i myszy [ 186 ]. Oprócz tego w ramach innego badania udało się z powodzeniem opracować transgeniczny model marmozety choroby Huntingtona (HD) [ 187 ]. Te marmozety wykazywały dystonię i pląsawicę – formy mimowolnych ruchów, które są fizjologicznymi fenotypami HD. Biorąc pod uwagę te wyniki, cenne może być kontynuowanie badań NDD na transgenicznych modelach NHP w ramach badań oceniających skuteczność terapii terapeutycznych w różnych punktach czasowych rozwoju neurorozwojowego.
Kolejną przeszkodą w przełożeniu klinicznym jest optymalizacja metod dostarczania leków. Rozważając zastosowanie wektorów wirusowych w NDD, należy wziąć pod uwagę zrównoważenie specyficzności tkankowej, immunogenności, ograniczeń pakowania i zdolności do penetracji BBB. Wraz z pojawieniem się technologii, takich jak wektory oparte na lipidach, możliwe będzie pokonanie przeszkód związanych z wektorami wirusowymi w opracowywaniu środków terapeutycznych. Ponieważ jednak wektory oparte na lipidach są wciąż w powijakach, potrzebne są dalsze badania, aby wyraźnie porównać zarówno skuteczność, jak i bezpieczeństwo wektorów wirusowych i lipidowych.
Bibliografia
-
Lord C, Elsabbagh M, Baird G, Veenstra-Vanderweele J. Zaburzenie ze spektrum autyzmu. Lancet 2018;392:508–20.
-
Lai MC, Kassee C, Besney R, Bonato S, Hull L, Mandy W i in. Częstość współwystępujących diagnoz zdrowia psychicznego w populacji autystycznej: przegląd systematyczny i metaanaliza. Lancet Psychiatry 2019;6:819–29.
-
Maenner MJ, Shaw KA, Bakian AV, Bilder DA, Durkin MS, Esler A i in. Częstość występowania i charakterystyka zaburzeń ze spektrum autyzmu wśród dzieci w wieku 8 lat – Sieć monitorowania autyzmu i niepełnosprawności rozwojowej, 11 miejsc, Stany Zjednoczone, 2018. MMWR Surveill Summ 2021;70:1–16.
-
Buescher AV, Cidav Z, Knapp M, Mandell DS. Koszty zaburzeń ze spektrum autyzmu w Wielkiej Brytanii i Stanach Zjednoczonych. JAMA Pediatr 2014;168:721–8.
-
Hartley SL, Barker ET, Seltzer MM, Greenberg JS, Floyd FJ. Satysfakcja małżeńska a doświadczenia rodzicielskie matek i ojców młodzieży i dorosłych z autyzmem. Am J Intellect Dev Disabil 2011;116:81–95.
-
Blaxill M, Rogers T, Nevison C. Tsunami autyzmu: wpływ rosnącej częstości występowania na społeczne koszty autyzmu w Stanach Zjednoczonych. J. Autyzm Dev Disord 2022;52:2627–43.
-
Sandin S, Lichtenstein P, Kuja-Halkola R, Hultman C, Larsson H, Reichenberg A. Dziedziczność zaburzeń ze spektrum autyzmu. JAMA 2017;318:1182–4.
-
Zespół Buitinga K, Williamsa C, Horsthemke B. Angelmana – spojrzenie na rzadkie zaburzenie neurogenetyczne. Nat Rev Neurol 2016;12:584–93.
-
Hagerman RJ, Berry-Kravis E, Hazlett HC, Bailey DB Jr, Moine H, Kooy RF i in. Zespół łamliwego chromosomu X. Nat Rev Dis Prim 2017;3:17065.
-
Sandweiss AJ, Brandt VL, Zoghbi HY. Postępy w zrozumieniu zespołu Retta i zespołu duplikacji MECP2: perspektywy przyszłych terapii. Lancet Neurol 2020;19:689–98.
-
Geschwind DH. Autyzm: wiele genów, wspólne ścieżki? Komórka 2008;135:391–5.
-
Iakoucheva LM, Muotri AR, Sebat J. Dotarcie do rdzeni autyzmu. Komórka 2019;178:1287–98.
-
Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T i in. Silny związek mutacji liczby kopii de novo z autyzmem. Nauka 2007;316:445–9.
-
Marshall CR, Noor A, Vincent JB, Lionel AC, Feuk L, Skaug J i in. Strukturalna zmienność chromosomów w zaburzeniach ze spektrum autyzmu. Am J Hum Genet 2008; 82: 477–88.
-
Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R i in. Funkcjonalny wpływ globalnej zmienności liczby rzadkich kopii w zaburzeniach ze spektrum autyzmu. Natura 2010;466:368–72.
-
Sanders SJ, Ercan-Sencicek AG, Hus V, Luo R, Murtha MT, Moreno-De-Luca D i in. Wiele nawracających CNV de novo, w tym duplikacje regionu zespołu Williamsa 7q11.23, są silnie powiązane z autyzmem. Neuron 2011;70:863–85.
-
Levy D, Ronemus M, Yamrom B, Lee YH, Leotta A, Kendall J i in. Rzadkie zmiany liczby kopii de novo i przekazywane w zaburzeniach ze spektrum autyzmu. Neuron 2011;70:886–97.
-
Weiss LA, Shen Y, Korn JM, Arking DE, Miller DT, Fossdal R i in. Związek między mikrodelecją i mikroduplikacją w 16p11.2 a autyzmem. N. Engl J Med 2008;358:667–75.
-
Antaki D, Guevara J, Maihofer AX, Klein M, Gujral M, Grove J i in. Fenotypowe spektrum autyzmu można przypisać połączonemu działaniu rzadkich wariantów, ryzyka wielogenowego i płci. Nat Genet. 2022;54:1284–92.
-
Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J i in. Zakłócenia genów de novo u dzieci ze spektrum autyzmu. Neuron 2012;74:285–99.
-
Neale BM, Kou Y, Liu L, Ma’ayan A, Samocha KE, Sabo A i in. Wzorce i wskaźniki egzonicznych mutacji de novo w zaburzeniach ze spektrum autyzmu. Natura 2012;485:242–5.
-
O’Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP i in. Sporadyczne egzomy autystyczne ujawniają wysoce połączoną sieć białek mutacji de novo. Natura 2012;485:246–50.
-
Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ i in. Mutacje de novo ujawnione w wyniku sekwencjonowania całego egzomu są silnie powiązane z autyzmem. Natura 2012;485:237–41.
-
De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE i in. Geny synaptyczne, transkrypcyjne i chromatyny zaburzone w autyzmie. Natura 2014;515:209–15.
-
Satterstrom FK, Kosmicki JA, Wang J, Breen MS, De Rubeis S, An JY i in. Zakrojone na szeroką skalę badanie sekwencjonowania egzomów implikuje zmiany rozwojowe i funkcjonalne w neurobiologii autyzmu. Komórka 2020;180:568–84 e23.
-
Fu JM, Satterstrom FK, Peng M, Brand H, Collins RL, Dong S i in. Rzadkie różnice w kodowaniu zapewniają wgląd w architekturę genetyczną i kontekst fenotypowy autyzmu. Nat Genet. 2022;54:1320–31.
-
O’Donnell-Luria AH, Pais LS, Faundes V, Wood JC, Sveden A, Luria V i in. Warianty heterozygotyczne w KMT2E powodują spektrum zaburzeń neurorozwojowych i padaczkę. Am J Hum Genet 2019; 104: 1210–22.
-
Gallagher D, Voronova A, Zander MA, Cancino GI, Bramall A, Krause MP i in. Ankrd11 to regulator chromatyny zaangażowany w autyzm, który jest niezbędny do rozwoju neuronów. Komórka deweloperska 2015;32:31–42.
-
Jung EM, Moffat JJ, Liu J, Dravid SM, Gurumurthy CB, Kim WY. Haploinsuficiency Arid1b zakłóca rozwój interneuronów korowych i zachowanie myszy. Nat Neurosci 2017;20:1694–707.
-
Katayama Y, Nishiyama M, Shoji H, Ohkawa Y, Kawamura A, Sato T i in. Haploinsuficiency CHD8 skutkuje fenotypami autystycznymi u myszy. Natura 2016;537:675–9.
-
Clipperton-Allen AE, strona DT. Myszy z haploinsuficientem Pten wykazują szeroki przerost mózgu, ale selektywne upośledzenia w testach behawioralnych istotnych dla autyzmu. Hum Mol Genet 2014;23:3490–505.
-
Peca J, Feliciano C, Ting JT, Wang W, Wells MF, Venkatraman TN i in. Myszy z mutacją Shank3 wykazują zachowania autystyczne i dysfunkcję prążkowia. Natura 2011;472:437–42.
-
Raveau M, Shimohata A, Amano K, Miyamoto H, Yamakawa K. Haploinsuficiency DYRK1A u myszy powoduje cechy autystyczne i drgawki gorączkowe. Neurobiol Dis 2018;110:180–91.
-
Amar M, Pramod AB, Yu NK, Herrera VM, Qiu LR, Moran-Losada P i in. Powiązana z autyzmem haploinsuficiencja linii zarodkowej Cullin3 wpływa na dynamikę cytoszkieletu i neurogenezę korową poprzez sygnalizację RhoA. Mol Psychiatria. 2021;26:3586–613.
-
Chen Y, Yu J, Niu Y, Qin D, Liu H, Li G i in. Modelowanie zespołu Retta przy użyciu zmutowanych małp Cynomolgus MECP2 poddanych edycji TALEN. Komórka 2017;169:945–55 e10.
-
Zhou Y, Sharma J, Ke Q, Landman R, Yuan J, Chen H i in. Nietypowe zachowanie i łączność u makaków z mutacją SHANK3. Natura 2019;570:326–31.
-
Grove J, Ripke S, Als TD, Mattheisen M, Walters RK, Won H i in. Identyfikacja typowych wariantów ryzyka genetycznego zaburzeń ze spektrum autyzmu. Nat Genet 2019;51:431–44.
-
Dunbar CE, High KA, Joung JK, Kohn DB, Ozawa K, Sadelain M. Terapia genowa wchodzi w wiek. Nauka 2018;359:6372.
-
Benger M, Kinali M, Mazarakis ND. Zaburzenie ze spektrum autyzmu: perspektywy leczenia za pomocą terapii genowej. Mol Autyzm 2018;9:39.
-
Rett A. O nietypowym zespole zaniku mózgu w przebiegu hiperamonemii w dzieciństwie. Wien Med Wochenschr 1966;116:723–6.
-
Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Zespół Retta jest spowodowany mutacjami w połączonym z X MECP2, kodującym białko wiążące metylo-CpG 2. Nat Genet 1999;23:185–8.
-
Sztainberg Y, Chen HM, Swann JW, Hao S, Tang B, Wu Z i in. Odwrócenie fenotypów u myszy z duplikacją MECP2 przy użyciu ratowania genetycznego lub oligonukleotydów antysensownych. Natura 2015;528:123–6.
-
Gadalla KK, Bailey ME, Spike RC, Ross PD, Woodard KT, Kalburgi SN i in. Lepsze przeżycie i zmniejszone nasilenie fenotypu po przeniesieniu genu AAV9/MECP2 na noworodkowe i młode samce myszy z nokautem Mecp2. Mol Ther 2013;21:18–30.
-
Luoni M, Giannelli S, Indrigo MT, Niro A, Massimino L, Iannielli A i in. Dostarczenie do całego mózgu podatnego na niestabilność transgenu Mecp2 poprawia behawioralne i molekularne defekty patologiczne w mysich modelach zespołu Retta. Elife 2020;9:e52629.
-
Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A i in. Identyfikacja genu (FMR-1) zawierającego powtórzenie CGG zbieżne z regionem klastra punktu przerwania wykazującym zmienność długości w zespole łamliwego chromosomu X. Komórka 1991;65:905–14.
-
Pieretti M, Zhang FP, Fu YH, Warren ST, Oostra BA, Caskey CT i in. Brak ekspresji genu FMR-1 w zespole łamliwego chromosomu X. Komórka 1991;66:817–22.
-
Gholizadeh S, Arsenault J, Xuan IC, Pacey LK, Hampson DR. Zmniejszone nasilenie fenotypu po dostarczeniu genu Fmr1 za pośrednictwem wirusa związanego z adenowirusem u myszy z zespołem łamliwego chromosomu X. Neuropsychofarmakologia 2014;39:3100–11.
-
Shaberman B. Organizacja non-profit zajmująca się badaniami nad siatkówką toruje drogę do komercjalizacji terapii genowych. Hum Gene Ther 2017;28:1118–21.
-
Darrow JJ. Luxturna: Dokumenty FDA ujawniają wartość kosztownej terapii genowej. Drug Discov Today 2019;24:949–54.
-
McKenzie AT, Wang M, Hauberg ME, Fullard JF, Kozlenkov A, Keenan A i in. Architektury sieciowe ekspresji i koekspresji genów specyficzne dla typu komórek mózgowych. Republika Naukowa 2018;8:8868.
-
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. Programowalna endonukleaza DNA kierowana podwójnym RNA w adaptacyjnej odporności bakteryjnej. Nauka 2012;337:816–21.
-
Pelechano V, Steinmetz LM. Regulacja genów poprzez transkrypcję antysensowną. Nat Rev Genet 2013;14:880–93.
-
Vanhée-Brossollet C, Vaquero C. Czy naturalne transkrypty antysensowne mają sens u eukariontów? Gene 1998;211:1–9.
-
Faghihi MA, Wahlestedt C. Role regulacyjne naturalnych transkryptów antysensownych. Nat Rev Mol Cell Biol 2009;10:637–43.
-
Wight M, Werner A. Funkcje naturalnych transkryptów antysensownych. Eseje Biochem 2013;54:91–101.
-
Shearwin KE, Callen BP, Egan JB. Interferencja transkrypcyjna – kurs przyspieszony. Trendy Genet 2005;21:339–45.
-
Liu Y, Liu H, Titus L, Boden SD. Naturalne transkrypty antysensowne wspomagają tworzenie kości poprzez zwiększenie sensownej transkrypcji IFITM5. Kość 2012;51:933–8.
-
Wolter JM, Mao H, Fragola G, Simon JM, Krantz JL, Bazick HO i in. Terapia genowa Cas9 w przypadku zespołu Angelmana zatrzymuje długie niekodujące RNA Ube3a-ATS. Przyroda 2020;587:281–4.
-
Meng L, Ward AJ, Chun S, Bennett CF, Beaudet AL, Rigo F. W kierunku terapii zespołu Angelmana poprzez celowanie w długi niekodujący RNA. Natura 2015;518:409–12.
-
Schmid RS, Deng X, Panikker P, Msackyi M, Breton C, Wilson JM. CRISPR/Cas9 skierowany na antysensowny transkrypt Ube3a poprawia fenotyp zespołu Angelmana u myszy. J Clin Invest. 2021;131:5.
-
Xie N, Gong H, Suhl JA, Chopra P, Wang T, Warren ST. Reaktywacja FMR1 przez delecję za pośrednictwem CRISPR/Cas9 rozszerzonego powtórzenia CGG delikatnego chromosomu X. PLoS One 2016;11:e0165499.
-
den Hollander AI, Koenekoop RK, Yzer S, Lopez I, Arends ML, Voesenek KE i in. Mutacje w genie CEP290 (NPHP6) są częstą przyczyną wrodzonej ślepoty Lebera. Am J Hum Genet 2006; 79: 556–61.
-
Maeder ML, Stefanidakis M, Wilson CJ, Baral R, Barrera LA, Bounoutas GS i in. Opracowanie metody edycji genów w celu przywrócenia utraty wzroku u pacjentów z wrodzoną ślepotą Lebera typu 10. Nat Med 2019;25:229–33.
-
Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE i in. Modularna regulacja transkrypcji u eukariontów za pośrednictwem CRISPR. Komórka 2013;154:442–51.
-
Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP i in. Zmiana przeznaczenia CRISPR na platformę kierowaną przez RNA do specyficznej dla sekwencji kontroli ekspresji genów. Komórka 2013;152:1173–83.
-
Dominguez AA, Lim WA, Qi LS. Poza edycją: ponowne wykorzystanie CRISPR-Cas9 do precyzyjnej regulacji genomu i przesłuchań. Nat Rev Mol Cell Biol 2016;17:5–15.
-
Sander JD, Joung JK. Systemy CRISPR-Cas do edycji, regulacji i namierzania genomów. Nat Biotechnol 2014;32:347–55.
-
Chavez A, Scheiman J, Vora S, Pruitt BW, Tuttle M, Iyer EPR i in. Wysoce wydajne programowanie transkrypcyjne za pośrednictwem Cas9. Metody Nat 2015;12:326–8.
-
Tanenbaum ME, Gilbert LA, Qi LS, Weissman JS, Vale RD. System znakowania białek do wzmacniania sygnału w ekspresji genów i obrazowaniu fluorescencyjnym. Komórka 2014;159:635–46.
-
Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C i in. Aktywacja transkrypcji w skali genomu przez opracowany kompleks CRISPR-Cas9. Natura 2015;517:583–8.
-
Chavez A, Tuttle M, Pruitt BW, Ewen-Campen B, Chari R, Ter-Ovanesyan D i in. Porównanie aktywatorów Cas9 u wielu gatunków. Metody Nat 2016;13:563–7.
-
Claes L, Del-Favero J, Ceulemans B, Lagae L, Van Broeckhoven C, De Jonghe P. Mutacje de novo w genie kanału sodowego SCN1A powodują ciężką padaczkę miokloniczną u niemowląt. Am J Hum Genet 2001; 68: 1327–32.
-
Sugawara T, Mazaki-Miyazaki E, Fukushima K, Shimomura J, Fujiwara T, Hamano S i in. Częste mutacje SCN1A w ciężkiej padaczce mioklonicznej w okresie niemowlęcym. Neurologia 2002;58:1122–4.
-
Hsiao J, Yuan TY, Tsai MS, Lu CY, Lin YC, Lee ML i in. Zwiększenie ekspresji genów haploinsuficient w mózgu poprzez celowanie w długi niekodujący RNA poprawia fenotyp napadów w modelu zespołu Dravet. EBioMedicine 2016;9:257–77.
-
Colasante G, Lignani G, Brusco S, Di Berardino C, Carpenter J, Giannelli S i in. Aktywacja genu Scn1a oparta na dCas9 przywraca hamującą pobudliwość interneuronów i łagodzi drgawki u myszy z zespołem Dravet. Mol Ther 2020;28:235–53.
-
Wolff M, Johannesen KM, Hedrich UBS, Masnada S, Rubboli G, Gardella E i in. Różnorodność genetyczna i fenotypowa sugeruje implikacje terapeutyczne w zaburzeniach związanych z SCN2A. Mózg 2017;140:1316–36.
-
Spratt PWE, Ben-Shalom R, Keeshen CM, Burke KJ Jr, Clarkson RL, Sanders SJ i in. Gen Scn2a związany z autyzmem przyczynia się do pobudliwości dendrytycznej i funkcji synaptycznej w korze przedczołowej. Neuron 2019;103:673–85.
-
Tamura S, AD Nelson, P Spratt, H. Kyoung, X Zhou, Z Li i in. Aktywacja CRISPR ratuje nieprawidłowości w zaburzeniu ze spektrum autyzmu związanym z haploinsufficiency SCN2A. 2022, https://doi.org/10.1101/2022.03.30.486483 .
-
Colasante G, Qiu Y, Massimino L, Di Berardino C, Cornford JH, Snowball A i in. In vivo CRISPRa zmniejsza napady padaczkowe i ratuje deficyty poznawcze w modelu padaczki u gryzoni. Mózg 2020;143:891–905.
-
Matharu N, Rattanasopha S, Tamura S, Maliskova L, Wang Y, Bernard A i in. Aktywacja promotora lub wzmacniacza za pośrednictwem CRISPR ratuje otyłość spowodowaną haploinsufficiency. Nauka 2019;363:6424.
-
Hunt C, Hartford SA, White D, Pefanis E, Hanna T, Herman C i in. Specyficzna tkankowo aktywacja ekspresji genów przez system CRISPRa Synergistycznego Mediatora Aktywacji (SAM) u myszy. Nat Commun 2021;12:2770.
-
Liang XH, Shen W, Crooke ST. Specyficzne zwiększenie poziomu białek poprzez wzmocnienie translacji przy użyciu antysensownych oligonukleotydów ukierunkowanych na otwarte ramki w górę. Adv Exp Med Biol 2017;983:129–46.
-
Meijer HA, Thomas AA. Kontrola syntezy białek eukariotycznych przez otwarte ramki odczytu znajdujące się powyżej w nieulegającym translacji regionie 5′ mRNA. Biochem J. 2002;367:1–11.
-
Liang XH, Shen W, Sun H, Migawa MT, Vickers TA, Crooke ST. Wydajność translacji mRNA jest zwiększana przez antysensowne oligonukleotydy ukierunkowane na otwarte ramki odczytu znajdujące się powyżej. Nat Biotechnol 2016;34:875–80.
-
Morris DR, Geballe AP. Upstream otwarte ramki odczytu jako regulatory translacji mRNA. Mol Cell Biol 2000;20:8635–42.
-
Kozak M. Okoliczności i mechanizmy hamowania translacji przez strukturę drugorzędową w eukariotycznych mRNA. Mol Cell Biol 1989;9:5134–42.
-
Walijski MJ, Smith AE. Molekularne mechanizmy dysfunkcji kanału chlorkowego CFTR w mukowiscydozie. Komórka 1993;73:1251–4.
-
Sasaki S, Sun R, Bui HH, Crosby JR, Monia BP, Guo S. Steric Hamowanie elementów regulacyjnych 5 'UTR powoduje zwiększenie regulacji ludzkiego CFTR. Mol Ther 2019;27:1749–57.
-
Cartegni L., Krainer AR. Zakłócenie zależnego od SF2/ASF wzmacniacza splicingu egzonowego w SMN2 powoduje rdzeniowy zanik mięśni przy braku SMN1. Nat Genet 2002;30:377–84.
-
Muntoni F., Wood MJ. Celowanie w RNA w leczeniu chorób nerwowo-mięśniowych. Nat Rev Drug Discov 2011;10:621–37.
-
Hua Y, Vickers TA, Baker BF, Bennett CF, Krainer AR. Wzmocnienie włączenia eksonu 7 SMN2 przez antysensowne oligonukleotydy ukierunkowane na ekson. PLoS Biol 2007;5:e73.
-
Kole R., Krieg AM. Terapia z pominięciem eksonu w dystrofii mięśniowej Duchenne’a. Adv Drug Deliv Rev 2015; 87: 104–7.
-
Prakash V. Spinraza – historia sukcesu choroby rzadkiej. Gene Ther 2017;24:497.
-
Mercuri E, Darras BT, Chiriboga CA, Day JW, Campbell C, Connolly AM i in. Nusinersen a kontrola pozorowana w rdzeniowym zaniku mięśni o późniejszym początku. N. Engl J Med 2018;378:625–35.
-
Finkel RS, Mercuri E, Darras BT, Connolly AM, Kuntz NL, Kirschner J i in. Nusinersen a kontrola pozorowana w rdzeniowym zaniku mięśni o początku dziecięcym. N. Engl J Med 2017;377:1723–32.
-
Malhotra SB, Hart KA, Klamut HJ, Thomas NS, Bodrug SE, Burghes AH i in. Delecje przesunięcia ramki u pacjentów z dystrofią mięśniową Duchenne’a i Beckera. Nauka 1988;242:755–9.
-
Echevarría L, Aupy P, Goyenvalle A. Postępy w pomijaniu egzonów w przypadku dystrofii mięśniowej Duchenne’a. Hum Mol Genet 2018;27:R163 – R172.
-
Voit T, Topaloglu H, Straub V, Muntoni F, Deconinck N, Campion G i in. Bezpieczeństwo i skuteczność drisapersenu w leczeniu dystrofii mięśniowej Duchenne’a (DEMAND II): eksploracyjne, randomizowane, kontrolowane placebo badanie fazy 2. Lancet Neurol 2014;13:987–96.
-
Flanigan KM, Voit T, Rosales XQ, Servais L, Kraus JE, Wardell C i in. Farmakokinetyka i bezpieczeństwo pojedynczych dawek drisapersenu u niechodzących pacjentów z dystrofią mięśniową Duchenne’a: wyniki randomizowanego badania klinicznego z podwójnie ślepą próbą. Zaburzenia nerwowo-mięśniowe 2014;24:16–24.
-
Goemans N, Mercuri E, Belousova E, Komaki H, Dubrovsky A, McDonald CM i in. Randomizowane, kontrolowane placebo badanie fazy 3 antysensownego oligonukleotydu, drisapersenu, w dystrofii mięśniowej Duchenne’a. Zaburzenia nerwowo-mięśniowe 2018;28:4–15.
-
Aartsma-Rus A, Krieg AM. FDA zatwierdza Eteplirsen w leczeniu dystrofii mięśniowej Duchenne’a: kolejny rozdział w sadze o Eteplirsenie. Kwas nukleinowy Ther 2017;27:1–3.
-
Batten FE, Mayo MS. Rodzinne zwyrodnienie mózgu ze zmianami w plamce żółtej. Proc R Soc Med 1915; 8: 70–90.
-
Dyken PR. Ponowne rozważenie klasyfikacji neuronalnych ceroidolipofuscynoz. Am J Med Genet Suppl 1988; 5: 69–84.
-
Kim J, Hu C, Moufawad El Achkar C, Black LE, Douville J, Larson A i in. Dostosowana do pacjenta terapia oligonukleotydowa w przypadku rzadkich chorób genetycznych. N. Engl J Med 2019;381:1644–52
-
Nomakuchi TT, Rigo F, Aznarez I, Krainer AR. Ukierunkowane na antysensowne oligonukleotydy hamowanie rozpadu mRNA za pośrednictwem nonsensu. Nat Biotechnol 2016;34:164–6.
-
Calvo SE, Pagliarini DJ, Mootha VK. Otwarte ramki odczytu poprzedzające powodują powszechne zmniejszenie ekspresji białek i są polimorficzne wśród ludzi. Proc Natl Acad Sci USA 2009;106:7507–12.
-
Lee S, Liu B, Huang SX, Shen B, Qian SB. Globalne mapowanie miejsc inicjacji translacji w komórkach ssaków z rozdzielczością pojedynczego nukleotydu. Proc Natl Acad Sci Usa 2012;109:E2424–32.
-
Walder RY, Walder JA. Rola RNazy H w translacji zatrzymanej hybrydowo przez oligonukleotydy antysensowne. Proc Natl Acad Sci USA 1988; 85: 5011–5.
-
Cheah CS, Yu FH, Westenbroek RE, Kalume FK, Oakley JC, Potter GB i in. Specyficzna delecja kanałów sodowych NaV1.1 w interneuronach hamujących powoduje drgawki i przedwczesną śmierć w mysim modelu zespołu Dravet. Proc Natl Acad Sci USA 2012;109:14646–51.
-
Milazzo C, Mientjes EJ, Wallaard I, Rasmussen SV, Erichsen KD, Kakunuri T i in. Leczenie oligonukleotydami antysensownymi ratuje ekspresję UBE3A i wiele fenotypów mysiego modelu zespołu Angelmana. Wgląd JCI. 2021;6:15.
-
Davidson BL, Gao G, Berry-Kravis E, Bradbury AM, Bonnemann C, Buxbaum JD i in. Terapia genowa rzadkich genetycznych zaburzeń neurorozwojowych. Mol Ther 2022;30:2416–28.
-
Cox DBT, Gootenberg JS, Abudayyeh OO, Franklin B, Kellner MJ, Joung J i in. Edycja RNA za pomocą CRISPR-Cas13. Nauka 2017;358:1019–27.
-
Marina RJ, Brannan KW, Dong KD, Yee BA, Yeo GW. Ocena opracowanych systemów za pośrednictwem CRISPR-Cas do edycji RNA specyficznej dla miejsca. Cell Rep. 2020;33:108350.
-
Sinnamon JR, Jacobson ME, Yung JF, Fisk JR, Jeng S, McWeeney SK i in. Ukierunkowana edycja RNA w pniu mózgu łagodzi dysfunkcje oddechowe w mysim modelu zespołu Retta. Proc Natl Acad Sci USA 2022;119:e2206053119.
-
Crino PB, Nathanson KL, Henske EP. Kompleks stwardnienia guzowatego. N. Engl J Med 2006;355:1345–56.
-
Liang N, Zhang C, Dill P, Panasyuk G, Pion D, Koka V i in. Regulacja YAP przez mTOR i autofagię ujawnia cel terapeutyczny kompleksu stwardnienia guzowatego. J Exp Med 2014;211:2249–63.
-
Salussolia CL, Klonowska K, Kwiatkowski DJ, Sahin M. Etiologie genetyczne, diagnostyka i leczenie zespołu stwardnienia guzowatego. Annu Rev Genomics Hum Genet 2019;20:217–40.
-
Tsai PT, Hull C, Chu Y, Greene-Colozzi E, Sadowski AR, Leech JM i in. Zachowania autystyczne i dysfunkcja móżdżku u myszy z mutacją Tsc1 w komórkach Purkinjego. Natura 2012;488:647–51.
-
Tsai PT, Rudolph S, Guo C, Ellegood J, Gibson JM, Schaeffer SM i in. Okresy wrażliwe na zachowania autystyczne za pośrednictwem móżdżku. Przedstawiciel komórki 2018;25:357–67.
-
Overwater IE, Rietman AB, Mous SE, Bindels-de Heus K, Rizopoulos D, Ten Hoopen LW i in. Randomizowane, kontrolowane badanie z ewerolimusem na IQ i autyzm w zespole stwardnienia guzowatego. Neurologia 2019;93:e200–e209.
-
Yizhar O, Fenno LE, Prigge M, Schneider F, Davidson TJ, O’Shea DJ i in. Równowaga pobudzenia/hamowania kory nowej w przetwarzaniu informacji i dysfunkcjach społecznych. Natura 2011;477:171–8.
-
Nelson SB, Valakh V. Równowaga pobudzająca / hamująca i homeostaza obwodów w zaburzeniach ze spektrum autyzmu. Neuron 2015;87:684–98.
-
Lee E, Lee J, Kim E. Brak równowagi pobudzenia/hamowania w zwierzęcych modelach zaburzeń ze spektrum autyzmu. Biol Psychiatria 2017;81:838–47.
-
Curia G, Papouin T, Séguéla P, Avoli M. Obniżenie poziomu tonicznego hamowania GABAergicznego w mysim modelu zespołu łamliwego chromosomu X. Kora Cereba 2009;19:1515–20.
-
Egawa K, Kitagawa K, Inoue K, Takayama M, Takayama C, Saitoh S i in. Zmniejszone hamowanie toniczne w komórkach ziarnistych móżdżku powoduje dysfunkcję motoryczną w mysim modelu zespołu Angelmana. Sci Transl Med 2012;4:163ra157.
-
Stoppel LJ, Kazdoba TM, Schaffler MD, Preza AR, Heynen A, Crawley JN i in. R-Baklofen odwraca deficyty poznawcze i poprawia interakcje społeczne w dwóch liniach myszy z delecją 16p11.2. Neuropsychofarmakologia 2018;43:513–24.
-
Roberts TPL, Bloy L, Blaskey L, Kuschner E, Gaetz L, Anwar A i in. Badanie MEG dotyczące ostrego podawania arbaklofenu (STX-209). Front Integr Neurosci 2019;13:69.
-
Berry-Kravis E, Hagerman R, Visootsak J, Budimirovic D, Kaufmann WE, Cherubini M i in. Arbaklofen w zespole łamliwego chromosomu X: wyniki badań III fazy. J Neurodev Disord 2017;9:3.
-
Niedźwiedź MF, Huber KM, Warren ST. Teoria mGluR upośledzenia umysłowego łamliwego chromosomu X. Trendy Neurosci 2004;27:370–7.
-
Silverman JL, Tolu SS, Barkan CL, Crawley JN. Powtarzające się zachowania związane z samopielęgnacją w mysim modelu autyzmu BTBR są blokowane przez antagonistę mGluR5, MPEP. Neuropsychofarmakologia 2010;35:976–89.
-
Mehta MV, Gandal MJ, Siegel SJ. Odwrócenie za pośrednictwem antagonisty mGluR5 podwyższonych stereotypowych, powtarzalnych zachowań w modelu autyzmu VPA. PLoS One 2011;6:e26077.
-
Tian D, Stoppel LJ, Heynen AJ, Lindemann L, Jaeschke G, Mills AA i in. Wkład mGluR5 w patofizjologię w mysim modelu mikrodelecji ludzkiego chromosomu 16p11.2. Nat Neurosci 2015;18:182–4.
-
Berry-Kravis E, Des Portes V, Hagerman R, Jacquemont S, Charles P, Visootsak J i in. Mavoglurant w zespole łamliwego chromosomu X: Wyniki dwóch randomizowanych, podwójnie ślepych badań kontrolowanych placebo. Sci Transl Med 2016;8:321ra5.
-
Howe JR, Bear MF, Golshani P, Klann E, Lipton SA, Mucke L i in. Mysz jako model rozwoju leków neuropsychiatrycznych. Curr Biol 2018;28:R909–R914.
-
Su T, Fan HX, Jiang T, Sun WW, Den WY, Gao MM i in. Wczesne ciągłe hamowanie sygnalizacji mGlu grupy 1 częściowo ratuje nieprawidłowości kręgosłupa dendrytycznego w mysim modelu z nokautem Fmr1 dla zespołu łamliwego chromosomu X. Psychopharmacol (Berl) 2011;215:291–300.
-
Azzarelli R, Kerloch T, Pacary E. Regulacja rozwoju kory mózgowej przez GTPazy Rho: spostrzeżenia z badań in vivo. Front Cell Neurosci 2014;8:445.
-
Escamilla CO, Filonova I, Walker AK, Xuan ZX, Holehonnur R, Espinosa F i in. Delecja Kctd13 zmniejsza transmisję synaptyczną poprzez zwiększoną RhoA. Natura 2017;551:227–31.
-
Richter M, Murtaza N, Scharrenberg R, White SH, Johanns O, Walker S i in. Zmieniona aktywność TAOK2 powoduje związane z autyzmem nieprawidłowości neurorozwojowe i poznawcze poprzez sygnalizację RhoA. Mol Psychiatria 2019;24:1329–50.
-
Urresti J, Zhang P, Moran-Losada P, Yu NK, Negraes PD, Trujillo CA i in. Organoidy korowe modelują wczesny rozwój mózgu zakłócony przez warianty liczby kopii 16p11.2 w autyzmie. Mol Psychiatria. 2021;26:7560–80.
-
Francis TC, Gaynor A, Chandra R, Fox ME, Lobo MK. Selektywny inhibitor RhoA, Rhosin, zwiększa odporność na stres poprzez zwiększenie plastyczności neuronów kolczastych D1 i zmniejszenie nadpobudliwości. Biol Psychiatria 2019;85:1001–10.
-
Kotterman MA, Chalberg TW, Schaffer DV. Wektory wirusowe w terapii genowej: perspektywy translacyjne i kliniczne. Annu Rev Biomed Eng 2015;17:63–89.
-
Shirley JL, de Jong YP, Terhorst C, Herzog RW. Odpowiedzi odpornościowe na wektory wirusowej terapii genowej. Mol Ther 2020;28:709–22.
-
Wang R, Wang R, Niu X, Cheng Y, Shang X, Li Y i in. Rola d-seryny pochodzącej z astrocytów w neurotoksyczności indukowanej PFOS przez NMDAR w pierwotnych neuronach hipokampa szczura. Toksykologia 2019;422:14–24.
-
Kumar M, Keller B, Makalou N, Sutton RE. Systematyczne wyznaczanie limitu pakowania wektorów lentiwirusowych. Hum Gene Ther 2001; 12: 1893–905.
-
Goswami R, Subramanian G, Silayeva L, Newkirk I, Doctor D, Chawla K i in. Terapia genowa pozostawia błędne koło. Front Oncol 2019;9:297.
-
Milone MC, O’Doherty U. Kliniczne zastosowanie wektorów lentiwirusowych. Białaczka 2018;32:1529–41.
-
Miller DG, Petek LM, Russell DW. Wektory wirusa związanego z adeno integrują się w miejscach złamania chromosomu. Nat Genet 2004;36:767–73.
-
Sabatino DE, Bushman FD, Chandler RJ, Crystal RG, Davidson BL, Dolmetsch R i in. Ocena stanu nauki w zakresie integracji wirusów związanych z adeno: zintegrowana perspektywa. Mol Ther 2022;30:2646–63.
-
Donsante A, Vogler C, Muzyczka N, Crawford JM, Barker J, Flotte T i in. Zaobserwowana częstość występowania nowotworów w długoterminowych badaniach wektorów rAAV na gryzoniach. Gene Ther 2001; 8: 1343–6.
-
Donsante A, Miller DG, Li Y, Vogler C, Brunt EM, Russell DW i in. Miejsca integracji wektorów AAV w mysim raku wątrobowokomórkowym. Nauka 2007;317:477.
-
Nguyen GN, Everett JK, Kafle S, Roche AM, Raymond HE, Leiby J i in. Długoterminowe badanie terapii genowej AAV u psów chorych na hemofilię A wykazało klonalną ekspansję transdukowanych komórek wątroby. Nat Biotechnol 2021;39:47–55.
-
Hanlon KS, Kleinstiver BP, Garcia SP, Zaborowski MP, Volak A, Spirig SE i in. Wysoki poziom integracji wektora AAV z pęknięciami DNA wywołanymi CRISPR. Nat Commun 2019;10:4439.
-
Nelson CE, Wu Y, Gemberling MP, Oliver ML, Waller MA, Bohning JD i in. Długoterminowa ocena edycji genomu AAV-CRISPR w przypadku dystrofii mięśniowej Duchenne’a. Nat Med 2019;25:427–32.
-
Nance E, Pun SH, Saigal R, Sprzedawcy DL. Dostarczanie leku do ośrodkowego układu nerwowego. Nat Rev Mater 2022;7:314–31.
-
Haery L, Deverman BE, Matho KS, Cetin A, Woodard K, Cepko C i in. Technologie i metody wirusów związanych z adenowirusami do ukierunkowanej manipulacji neuronowej. Front Neuroanat 2019;13:93.
-
Foust KD, Nurre E, Montgomery CL, Hernandez A, Chan CM, Kaspar BK. Wewnątrznaczyniowy AAV9 preferencyjnie celuje w neurony noworodków i astrocyty dorosłych. Nat Biotechnol 2009;27:59–65.
-
Day JW, Chiriboga CA, Crawford TO, Darras BT, Finkel RS, Connolly AM i in. AVXS-101 Terapia zastępcza genami (GRT) w leczeniu rdzeniowego zaniku mięśni typu 1 (SMA1): Aktualizacja kluczowego badania fazy 3 (STR1VE) (P1.6-058). Neurologia 2019;92:P1.6–058.
-
Mendell JR, Al-Zaidy S, Shell R, Arnold WD, Rodino-Klapac LR, Prior TW i in. Terapia zastępcza genami w pojedynczej dawce w leczeniu rdzeniowego zaniku mięśni. N. Engl J Med 2017;377:1713–22.
-
Ran FA, Cong L, Yan WX, Scott DA, Gootenberg JS, Kriz AJ i in. Edycja genomu in vivo przy użyciu Staphylococcus aureus Cas9. Natura 2015;520:186–91.
-
Friedland AE, Baral R, Singhal P, Loveluck K, Shen S, Sanchez M i in. Charakterystyka Staphylococcus aureus Cas9: mniejszy Cas9 do dostarczania wirusa związanego z adenowirusem typu „wszystko w jednym” i zastosowań sparowanych nickaz. Biol genomu 2015;16:257.
-
Bengtsson NE, Hall JK, Odom GL, Phelps MP, Andrus CR, Hawkins RD i in. Edycja genu dystrofiny CRISPR/Cas9 specyficznego dla mięśni poprawia patofizjologię w mysim modelu dystrofii mięśniowej Duchenne’a. Nat Commun 2017;8:14454.
-
Kim E, Koo T, Park SW, Kim D, Kim K, Cho HY i in. Edycja genomu in vivo za pomocą małego ortologu Cas9 pochodzącego z Campylobacter jejuni. Nat Commun 2017;8:14500.
-
Deverman BE, Pravdo PL, Simpson BP, Kumar SR, Chan KY, Banerjee A i in. Selekcja zależna od Cre daje warianty AAV umożliwiające powszechny transfer genów do mózgu dorosłego. Nat Biotechnol 2016;34:204–9.
-
Morabito G, Giannelli SG, Ordazzo G, Bido S, Castoldi V, Indrigo M i in. Ekspresja na skalę globalną za pośrednictwem AAV-PHP.B w układzie nerwowym myszy umożliwia terapię genową GBA1 w celu zapewnienia szerokiej ochrony przed synukleinopatią. Mol Ther 2017;25:2727–42.
-
Chan KY, Jang MJ, Yoo BB, Greenbaum A, Ravi N, Wu WL i in. Zaprojektowane AAV do wydajnego, nieinwazyjnego dostarczania genów do centralnego i obwodowego układu nerwowego. Nat Neurosci 2017;20:1172–9.
-
Goertsen D, Flytzanis NC, Goeden N, Chuapoco MR, Cummins A, Chen Y i in. Warianty kapsydu AAV z ekspresją transgenu w całym mózgu i zmniejszonym ukierunkowaniem na wątrobę po podaniu dożylnym u myszy i marmozet. Nat Neurosci 2022;25:106–15.
-
Mathiesen SN, Lock JL, Schoderboeck L, Abraham WC, Hughes SM. Korzyści z transdukcji OUN dla AAV-PHP.eB w porównaniu z AAV9 zależą od drogi podawania i szczepu myszy. Metody Mol Ther Clin Dev 2020;19:447–58.
-
Yao Y, Wang J, Liu Y, Qu Y, Wang K, Zhang Y i in. Warianty serotypu 9 wirusa związanego z adenowirusem o zwiększonej penetracji bariery krew-mózg u gryzoni i naczelnych. Nat Biomed Eng 2022;6:1257–71.
-
Ma F, Yang L, Sun Z, Chen J, Rui X, Glass Z i in. Lipidoidy pochodzące z neuroprzekaźników (NT-lipidoidy) w celu zwiększenia dostarczania do mózgu poprzez wstrzyknięcie dożylne. Adv Sci 2020;6:eabb4429.
-
Gonzalez-Carter D, Liu X, Tockary TA, Dirisala A, Toh K, Anraku Y i in. Skierowanie nanocząstek do mózgu poprzez wykorzystanie nieprzepuszczalności bariery krew-mózg do selektywnego znakowania śródbłonka mózgu. Proc Natl Acad Sci Usa 2020;117:19141–50.
-
Roberts TC, Langer R, Wood MJA. Postępy w dostarczaniu leków oligonukleotydowych. Nat Rev Drug Discov 2020;19:673–94.
-
Hou X, Zaks T, Langer R, Dong Y. Nanocząsteczki lipidowe do dostarczania mRNA. Nat Rev Mater 2021;6:1078–94.
-
Gillmore JD, Gane E, Taubel J, Kao J, Fontana M, Maitland ML i in. Edycja genów CRISPR-Cas9 in vivo w przypadku amyloidozy transtyretynowej. N. Engl J Med. 2021;385:493–502.
-
Qiu M, Glass Z, Chen J, Haas M, Jin X, Zhao X i in. Wspólne dostarczanie mRNA Cas9 i RNA z pojedynczym przewodnikiem za pośrednictwem nanocząstek lipidowych pozwala na specyficzną dla wątroby edycję genomu Angptl3 in vivo. Proc Natl Acad Sci USA. 2021;118:10.
-
Glinos DA, Garborcauskas G, Hoffman P, Ehsan N, Jiang L, Gokden A i in. Zmienność transkryptomu w tkankach ludzkich ujawniona przez sekwencjonowanie o długim odczycie. Przyroda 2022;608:353–9.
-
Gandal MJ, Zhang P, Hadjimichael E, Walker RL, Chen C, Liu S i in. Rozregulowanie poziomu izoform obejmujące cały transkryptom w ASD, schizofrenii i chorobie afektywnej dwubiegunowej. Nauka 2018;362:6420.
-
Chau KK, Zhang P, Urresti J, Amar M, Pramod AB, Chen J i in. Pełnej długości transkryptom izoformy rozwijającego się ludzkiego mózgu dostarcza dalszych informacji na temat autyzmu. Cell Rep. 2021;36:109631.
-
Leung SK, Jeffries AR, Castanho I, Jordan BT, Moore K, Davies JP i in. Sekwencjonowanie pełnej długości transkryptów kory mózgowej człowieka i myszy pozwala zidentyfikować powszechną różnorodność izoform i alternatywny splicing. Cell Rep. 2021;37:110022.
-
Flaherty E, Zhu S, Barretto N, Cheng E, Deans PJM, Fernando MB i in. Wpływ neuronalny specyficznego dla pacjenta nieprawidłowego splicingu NRXN1alfa. Nat Genet 2019;51:1679–90.
-
Willsey AJ, Sanders SJ, Li M, Dong S, Tebbenkamp AT, Muhle RA i in. Sieci koekspresji wskazują, że ludzkie neurony projekcyjne głębokiej korowej połowy płodu są zaangażowane w patogenezę autyzmu. Komórka 2013;155:997–1007.
-
Parikshak NN, Luo R, Zhang A, Won H, Lowe JK, Chandran V i in. Integracyjne analizy genomu funkcjonalnego wskazują na specyficzne szlaki i obwody molekularne w autyzmie. Komórka 2013;155:1008–21.
-
Lin GN, Corominas R, Lemmens I, Yang X, Tavernier J, Hill DE i in. Przestrzenno-czasowa sieć białek 16p11.2 implikuje rozwój mózgu późnego połowy płodu w korze mózgowej i szlak KCTD13-Cul3-RhoA w chorobach psychicznych. Neuron 2015;85:742–54.
-
Silva-Santos S, van Woerden GM, Bruinsma CF, Mientjes E, Jolfaei MA, Distel B i in. Przywrócenie Ube3a identyfikuje odrębne okna rozwojowe w mysim modelu zespołu Angelmana. J Clin Invest 2015;125:2069–76.
-
Rotaru DC, van Woerden GM, Wallaard I, Elgersma Y. Przywrócenie genu Ube3a u dorosłych przywraca deficyty elektrofizjologiczne neuronów warstwy 5 kory przedczołowej w mysim modelu zespołu Angelmana. J Neurosci 2018;38:8011–30.
-
Guy J, Gan J, Selfridge J, Cobb S, Bird A. Odwrócenie wad neurologicznych w mysim modelu zespołu Retta. Nauka 2007;315:1143–7.
-
Kita Y, Nishibe H, Wang Y, Hashikawa T, Kikuchi SS, UM i in. Profilowanie ekspresji genów metodą rozdzielczości komórkowej w mózgu noworodków marmozet ujawnia dynamiczne różnice specyficzne dla gatunku i regionu. Proc Natl Acad Sci USA. 2021;118:18.
-
Yang SH, Cheng PH, Banta H, Piotrowska-Nitsche K, Yang JJ, Cheng EC i in. W kierunku transgenicznego modelu choroby Huntingtona u naczelnych innych niż człowiek. Natura. 2008;453:921–4.
Autorzy
Psychiatria translacyjna tom 13 , Numer artykułu: 58 ( 2023 )
Podziękowanie
Prace te zostały częściowo wsparte następującymi grantami: R01MH108528 (LMI), R01MH109885 (LMI), R21MH128827 (LMI), R56MH128365 (LMI) oraz grantami nr 345469 i 896503 Fundacji Simonsa na rzecz badań nad autyzmem dla LMI. Dziękujemy dr Josephowi Gleesonowi za przesłanie komentarzy na temat manuskryptu, a dr. Stephenowi Tranowi i Jonathanowi Sebatowi za pomocne dyskusje. Figury zostały stworzone za pomocą BioRender.com.
Deklaracje etyczne
Konkurencyjne interesy
Autorzy nie deklarują żadnych konkurencyjnych interesów.
Dodatkowe informacje
Nota wydawcy Springer Nature pozostaje neutralna w odniesieniu do roszczeń jurysdykcyjnych w publikowanych mapach i powiązaniach instytucjonalnych.
Prawa i uprawnienia
Otwarty dostęp Ten artykuł jest objęty licencją Creative Commons Uznanie autorstwa 4.0, która pozwala na używanie, udostępnianie, adaptację, dystrybucję i reprodukcję na dowolnym nośniku lub w dowolnym formacie, pod warunkiem odpowiedniego podania oryginalnego autora (autorów) i źródła, podaj link do licencji Creative Commons i wskaż, czy wprowadzono zmiany. Obrazy lub inne materiały stron trzecich zawarte w tym artykule są objęte licencją Creative Commons artykułu, chyba że w linii kredytowej dotyczącej materiału wskazano inaczej. Jeśli materiał nie jest objęty licencją Creative Commons artykułu, a zamierzone użycie jest niezgodne z przepisami ustawowymi lub przekracza dozwolone użycie, konieczne będzie uzyskanie zgody bezpośrednio od właściciela praw autorskich. Aby wyświetlić kopię tej licencji, odwiedź http://creativecommons.org/licenses/by/4.0/ .
Przedruki i pozwolenia
Link do artykułu: https://www.nature.com/articles/s41398-023-02356-y
O tym artykule


Zacytuj ten artykuł
Hong, D., Iakoucheva, LM Strategie terapeutyczne dla autyzmu: ukierunkowanie na trzy poziomy centralnego dogmatu biologii molekularnej. Transl Psychiatry 13 , 58 (2023). https://doi.org/10.1038/s41398-023-02356-y