


Chalkolityczna kopalnia miedzi w dolinie Timna, pustynia Negew, Izrael.
[Zamieszczony poniżej artykuł ma charakter bardzo specjalistyczny, naukowy, zdecydowaliśmy się jednak umieścić go na łamach WOBEC, gdyż opisywane w nim badania po raz kolejny dowodzą, że migracje ludności, przemieszczanie się ludów i kultur trwają od tysiącleci, odbywały się zawsze, są w dziejach ludzkości zjawiskiem powszechnym i stałym. Zmieniała się co najwyżej ich intensywność.
Redakcja]
Abstrakt
Kultura materialna późnego chalkolitu w południowym Lewancie (4500–3900 / 3800 p.n.e.) różni się jakościowo od poprzednich i późniejszych okresów. Tutaj, aby przetestować hipotezę, że na pojawienie się i upadek tej kultury wpłynęły ruchy ludzi, wygenerowaliśmy starożytny DNA obejmujący cały genom od 22 osób z jaskini Peqi’in w Izraelu. Osoby te były częścią jednorodnej populacji, którą można modelować jako wywodzącą ~ 57% swojego pochodzenia z grup spokrewnionych z lokalnym neolitem Lewantu, ~ 17% z grup spokrewnionych z irańskim chalkolitu i ~ 26% z grupy związane z neolitem anatolijskim. Wydaje się również, że populacja Peqi’in wniosła inny wkład do późniejszych grup z epoki brązu, z których jedna, jak pokazujemy, nie mogła prawdopodobnie pochodzić z tej samej populacji, co populacja jaskini Peqi’in.
s41467-018-05649-9Tłumaczenie
Wstęp
Kultura materialna późnego chalkolitu w południowym Lewancie kontrastuje jakościowo z kulturą z wcześniejszych i późniejszych okresów w tym samym regionie. Późny chalkolit w Lewancie charakteryzuje się wzrostem gęstości osadnictwa, wprowadzeniem sanktuariów 1 , 2 , 3 , wykorzystaniem ossuariów w pochówkach wtórnych 4 , 5 , a także ekspansją publicznych praktyk rytualnych oraz rozkwitem motywów symbolicznych rzeźbionych i malowane na artefaktach z ceramiki, bazaltu, miedzi i kości słoniowej 6 , 7 , 8 , 9. Imponujące metalowe artefakty z tego okresu, które odzwierciedlają pierwsze znane zastosowanie techniki „traconego wosku” do odlewania miedzi, świadczą o niezwykłych umiejętnościach technicznych ludzi tego okresu 10 , 11 .
Charakterystyczne cechy kulturowe późnego chalkolitu w Lewancie (często związane z kulturą Ghassulian, chociaż termin ten w praktyce nie jest stosowany w regionie Galilei, w którym opiera się niniejsza praca) mają niewiele stylistycznych powiązań z wcześniejszymi lub późniejszymi kulturami materialnymi regionu, co doprowadziło do szeroko zakrojonej debaty na temat pochodzenia ludzi, którzy stworzyli tę kulturę materialną. Jedna z hipotez głosi, że kultura chalkolitu w regionie została częściowo rozprzestrzeniona przez imigrantów z północy (tj. z północnej Mezopotamii), w oparciu o podobieństwa w projektach artystycznych 12 , 13. Inni sugerowali, że lokalna ludność Lewantu była całkowicie odpowiedzialna za rozwój tej kultury i że wszelkie podobieństwa do kultur materialnych na północy wynikają z zapożyczania idei, a nie z przemieszczania się ludzi 2 , 14 , 15 , 16 , 17 , 18 , 19.
Aby zbadać te pytania, zbadaliśmy starożytne DNA ze stanowiska chalkolitycznego w północnym Izraelu, Peqi’in (ryc. 1a ). Ta jaskinia, która ma około 17 m długości i 4,5–8,0 m szerokości (ryc. 1b ), została odkryta podczas budowy drogi w 1995 r. I została uszczelniona przez naturalne procesy podczas lub pod koniec późnego okresu chalkolitu (około 3900 pne) . Wykopaliska archeologiczne ujawniły niezwykłą gamę misternie wykonanych przedmiotów, w tym kielichy, misy i maselnice, a także ponad 200 ossuariów i dzbanów domowych, które zostały przekształcone w ossuaria (największa liczba, jaką kiedykolwiek znaleziono w jednej jaskini), często ozdobione antropomorficznymi wzorami (Rys. 1c ) 20 , 21. Oszacowano, że jaskinia grobowa zawierała do 600 osobników 22 , co czyni ją największym miejscem pochówku, jakie kiedykolwiek zidentyfikowano z okresu późnego chalkolitu w Lewancie. Bezpośrednie datowanie radiowęglowe sugeruje, że jaskinia była używana przez cały późny chalkolit (4500-3900 pne), funkcjonując jako centralne miejsce pochówku w regionie 21 , 23.


Tło witryny. a Lokalizacja miejsca, w którym znajduje się jaskinia Peqi’in. b Zdjęcie wnętrza jaskini Peqi’in. Zdjęcie Hila May, dzięki uprzejmości Centrum Ewolucji Człowieka i Biohistorii im. Dana Davida. c Zdjęcie kilku urn grobowych (ossuariów) z jaskini Peqi’in. Pasek skali: 10 cm. Zdjęcie Mariana Salzberger, dzięki uprzejmości Israel Antiquities Authority. Ta liczba nie jest objęta licencją CC BY dla tego artykułu. Wszelkie prawa zastrzeżone.
Wcześniejsze badania starożytnego DNA obejmujące cały genom z Bliskiego Wschodu ujawniły, że w czasie, gdy rozwijało się rolnictwo, populacje z Anatolii, Iranu i Lewantu były mniej więcej tak samo zróżnicowane genetycznie, jak dzisiejsi Europejczycy i mieszkańcy Azji Wschodniej 24 , 25. Jednak w epoce brązu ekspansja różnych bliskowschodnich populacji rolniczych — anatolijskich, irańskich i lewantyńskich — we wszystkich kierunkach i wzajemne mieszanie się zasadniczo ujednoliciła populacje w całym regionie, przyczyniając się w ten sposób do stosunkowo niskiego zróżnicowania genetycznego, które dominuje dzisiaj 24 . Lazaridis i in.24wykazało, że populacja Lewantu z epoki brązu z miejsca „Ain Ghazal w Jordanii (2490–2300 pne) może być statystycznie dopasowana jako mieszanka około 56% pochodzenia z grupy spokrewnionej z rolnikami z neolitu lewantyńskiego przed ceramiką (reprezentowane przez starożytne DNA z Motza w Izraelu i „Ain Ghazal w Jordanii; 8300–6700 pne) i 44% związanych z populacjami irańskiego chalkolitu (Seh Gabi, Iran; 4680–3662 kal. p.n.e.). Haber i in. 26zasugerował, że populacja kananejskiego Lewantu z epoki brązu z miejsca Sydonu w Libanie (~ 1700 p.n.e.) może być modelowana jako mieszanka tych samych dwóch grup, aczkolwiek w różnych proporcjach (48% związanych z neolitem Lewantu i 52% z irańskim chalkolitem) . Jednak dotychczas przeanalizowane stanowiska z epoki neolitu i epoki brązu w Lewancie są oddalone w czasie o ponad trzy tysiące lat, co sprawia, że badanie próbek wypełniających tę lukę, takich jak te z Peqi’in, ma kluczowe znaczenie.
W specjalnym pomieszczeniu czystym w Harvard Medical School uzyskaliśmy proszek kostny z 48 szczątków szkieletowych, z których 37 to kości skaliste znane z doskonałego zachowania DNA 27 . Wyekstrahowaliśmy DNA 28 i zbudowaliśmy biblioteki sekwencjonowania nowej generacji, do których dołączyliśmy unikalne kody kreskowe, aby zminimalizować możliwość zanieczyszczenia. Biblioteki potraktowaliśmy glikozylazą uracylowo-DNA (UDG), aby zredukować charakterystyczne starożytne uszkodzenia DNA, z wyjątkiem pierwszego i ostatniego nukleotydu 29 (tabela uzupełniająca 1 i dane uzupełniające 1zapewnić tło dla udanych próbek i podać informacje odpowiednio dla każdej biblioteki). Po wstępnym skriningu poprzez wzbogacenie bibliotek pod kątem mitochondrialnego DNA, wzbogaciliśmy obiecujące biblioteki o sekwencje pokrywające się z około 1,2 milionami polimorfizmów pojedynczego nukleotydu (SNP) 30 , 31. Oceniliśmy każdą osobę pod kątem autentyczności starożytnego DNA, ograniczając się do bibliotek z minimum 3% błędami cytozyny do tyminy na końcowym nukleotydzie 29, wymagając, aby stosunek sekwencji chromosomu X do Y był charakterystyczny dla obu samca lub samicę, wymagając >95% dopasowania do sekwencji konsensusowej mitochondrialnego DNA 30oraz wymagając (w przypadku samców) braku zmienności w znanych pozycjach polimorficznych na chromosomie X (szacunki punktowe zanieczyszczenia poniżej 2%) 32 . Ograniczyliśmy się również do osób, które przynajmniej raz objęły co najmniej 5000 docelowych SNP.
Ta procedura dała dane dotyczące całego genomu od 22 starożytnych osobników z jaskini Peqi’in (4500–3900 calBCE), przy czym osoby miały medianę 358 313 docelowych SNP objętych co najmniej raz (zakres: 25 171–1 002 682). Zbiór danych jest wyjątkowej jakości, biorąc pod uwagę zazwyczaj słabe zachowanie DNA na ciepłym Bliskim Wschodzie, z wyższym odsetkiem próbek dających znaczne pokrycie starożytnego DNA niż wcześniej uzyskano z regionu, prawdopodobnie odzwierciedlając zastosowane przez nas optymalne techniki pobierania próbek i dobre warunki konserwatorskie w jaskini. Przeanalizowaliśmy ten zestaw danych w połączeniu z wcześniej opublikowanymi zbiorami danych starożytnych populacji Bliskiego Wschodu 24 , 26 rzucić światło na historię osób pochowanych w jaskini Peqi’in oraz na dynamikę populacji Lewantu w okresie późnego chalkolitu.
Wyniki
Zróżnicowanie i różnorodność genetyczna w starożytnym Lewancie
W sumie 20 próbek Peqi’in wydaje się być niezwiązanych ze sobą w granicach naszej rozdzielczości (to znaczy analiza genetyczna sugeruje, że nie były one krewnymi pierwszego, drugiego ani trzeciego stopnia) i wykorzystaliśmy je jako nasz zestaw analiz. Korzystając z nowego punktu danych dodanego przez próbki Peqi’in, zaczęliśmy od zbadania, jak zróżnicowanie genetyczne między populacjami lewantyńskimi zmieniało się w czasie. Powtórzyliśmy poprzednie doniesienia o dramatycznym spadku zróżnicowania genetycznego w czasie w Zachodniej Eurazji 24 , obserwując medianę parami F ST wynoszącą 0, 023 (zakres: 0, 009–0, 061) między próbkami Peqi’in (skrót: Levant_ChL) a innymi zachodnio-euroazjatyckimi neolitami i chalkolitami populacji, w stosunku do wcześniej podanej mediany parami F ST0,098 (zakres: 0,023–0,153) obserwowany między populacjami w okresach przedneolitycznych, 0,015 (zakres: 0,002–0,045) w okresach epoki brązu i 0,011 (zakres: 0–0,046) we współczesnych populacjach zachodniej Eurazji 24 . Tak więc upadek do obecnych poziomów zróżnicowania został w dużej mierze zakończony przez chalkolit (rysunek uzupełniający 1 ).
Obserwujemy również wzrost różnorodności genetycznej w czasie w Lewancie, mierzony szybkością polimorfizmu między dwiema losowymi sekwencjami genomu w każdym SNP analizowanym w naszym badaniu. Konkretnie, populacja Levant_ChL wykazuje pośredni poziom heterozygotyczności względem wcześniejszych i późniejszych populacji (Fig. 2 ).

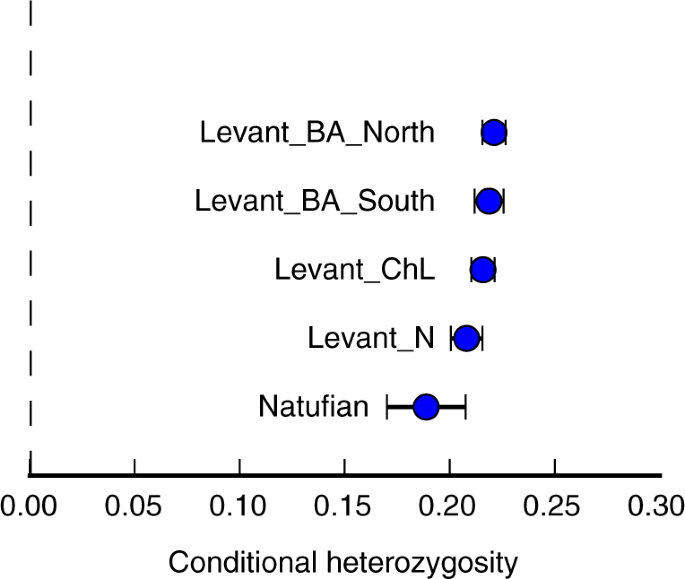
Różnorodność genetyczna w starożytnym Lewancie. Z biegiem czasu heterozygotyczność wzrasta w starożytnych populacjach lewantyńskich. Podano szacunkową statystykę ± 3 błędy standardowe.
Zarówno rosnąca różnorodność genetyczna w czasie, jak i zmniejszone zróżnicowanie między populacjami mierzone za pomocą F ST są zgodne z modelem, w którym przepływ genów zmniejszał zróżnicowanie między grupami, jednocześnie zwiększając różnorodność w obrębie grup.
Genetyczne podobieństwa osobników z jaskini Peqi’in
Aby uzyskać jakościowy obraz tego, jak te osoby odnoszą się do wcześniej opublikowanego starożytnego DNA i do współczesnych ludzi, zaczęliśmy od przeprowadzenia analizy głównych składowych (PCA) 33 . Na wykresie pierwszej i drugiej składowej głównej (ryc. 3a), próbki z jaskini Peqi’in tworzą ciasne skupisko, wspierając grupowanie tych osobników w pojedynczą populację do analizy (chociaż używamy szerokiej nazwy „Levant_ChL” w odniesieniu do tych próbek, zdajemy sobie sprawę, że są one obecnie jedynymi starożytnymi DNA dostępne z Lewantu w tym okresie i przyszłe prace prawdopodobnie ujawnią podstrukturę genetyczną w próbkach chalkolitu w szerokim regionie). Klaster Levant_ChL pokrywa się w PCA z klastrem zawierającym neolityczne próbki lewantyńskie (Levant_N), chociaż jest nieco przesunięty w górę na wykresie w kierunku klastra odpowiadającego próbkom z epoki brązu Lewantu, w tym próbkom z „Ain Ghazal, Jordania (Levant_BA_South) i Sydon w Libanie (Levant_BA_North).24 .

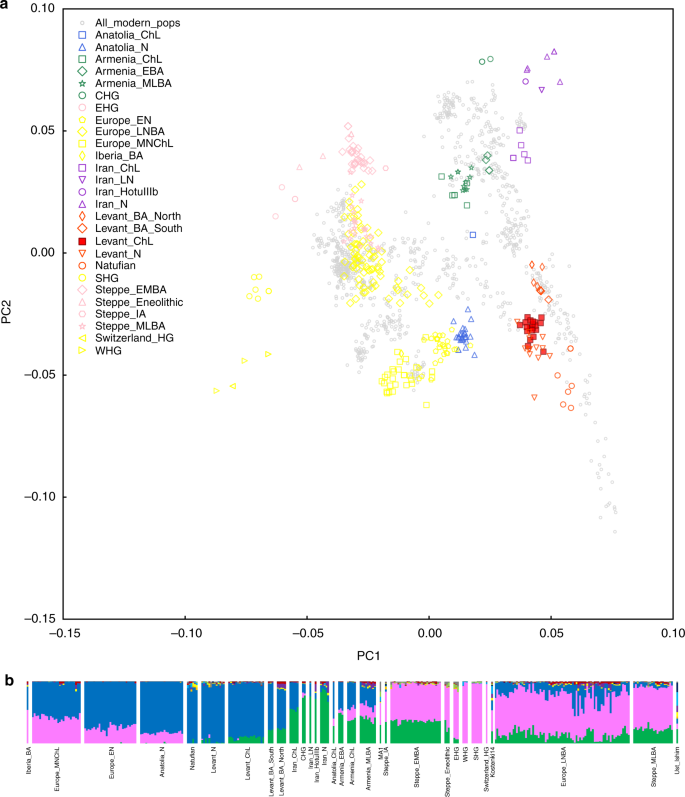
Struktura genetyczna analizowanych osobników. a Analiza głównych składowych 984 współczesnych mieszkańców Zachodniej Eurazji (pokazana na szaro) z 306 starożytnymi próbkami rzutowanymi na pierwsze dwie główne osie składowe i oznaczonymi według kultury. b Analiza DOMIESZANKI 984 i 306 starożytnych próbek z K = 11 składników przodków. Pokazane są tylko starożytne próbki
Analiza skupień oparta na modelu ADMIXTURE34 przyniosła wyniki zgodne z PCA, sugerując, że osobnicy z populacji Levant_ChL mieli średnio większe pokrewieństwo z irańskimi populacjami związanymi z rolnictwem niż miało to miejsce w przypadku wcześniejszych osobników lewantyńskich. Rysunek 3b pokazuje wyniki ADMIXTURE dla starożytnych osobników przy założeniu K = 11 klastrów (wybraliśmy tę liczbę, ponieważ maksymalizuje ona komponenty rodowe, które są skorelowane ze starożytnymi populacjami z Lewantu, z Iranu i europejskimi łowcami-zbieraczami)24. Podobnie jak w przypadku wszystkich populacji lewantyńskich, główny komponent rodowy przypisany do populacji Levant_ChL, pokazany na niebiesko, jest maksymalizowany u wcześniejszych osobników Levant_N i Natufian. ADMIXTURE przypisuje również składnik rodowodu w Levant_ChL, pokazany na zielono, do populacji, która jest ogólnie nieobecna we wcześniejszych populacjach Levant_N i Natufian, ale jest obecna w późniejszych próbkach Levant_BA_South i Levant_BA_North. Ten zielony komponent jest również wnioskowany w niewielkich proporcjach w kilku próbkach przypisanych do Levant_N, ale nie ma wyraźnego związku z lokalizacją archeologiczną lub datą, a osobniki te nie różnią się znacząco genetycznie od innych osobników włączonych do Levant_N przez formalne testy, a zatem łączymy wszystkie Levant_N dla głównych analiz w tym badaniu (Dodatkowa uwaga 1)24.
Ciągłość populacji i domieszki w Lewancie
Aby określić związek populacji Levant_ChL z innymi starożytnymi populacjami bliskowschodnimi, użyliśmy f-statystyki35 (więcej szczegółów w Supplementary Note 2). Najpierw oceniliśmy, czy populacja Levant_ChL jest zgodna z pochodzeniem bezpośrednio z populacji związanej z wcześniejszym Levant_N. Gdyby tak było, oczekiwalibyśmy, że populacja Levant_N byłaby zgodna z byciem bliżej spokrewnioną z populacją Levant_ChL niż z jakąkolwiek inną populacją, i rzeczywiście potwierdzamy to, obserwując dodatnią statystykę w postaci f4 (Levant_ChL, A; Levant_N, Chimpanzee) dla wszystkich starożytnych populacji testowych, A (ryc. 4a). Jednakże populacje Levant_ChL i Levant_N nie tworzą kladu, ponieważ gdy obliczamy statystykę symetrii formy f4 (Levant_N, Levant_ChL; A, Chimpanzee), stwierdzamy, że statystyka ta jest często ujemna, przy czym populacje bliskowschodnie spoza Lewantu dzielą więcej alleli z Levant_ChL niż z Levant_N (ryc. 4b). Wnioskujemy, że podczas gdy populacje Levant_N i Levant_ChL są wyraźnie spokrewnione, populacja Levant_ChL nie może być modelowana jako pochodząca bezpośrednio z populacji Levant_N bez dodatkowej domieszki związanej ze starożytnymi irańskimi rolnikami. Bezpośrednie dowody na to, że Levant_ChL jest domieszką pochodzą ze statystyki f3 (Levant_ChL; Levant_N, A), która dla niektórych populacji, A, jest znacząco ujemna, wskazując, że częstotliwości alleli w Levant_ChL mają tendencję do bycia pośrednimi między tymi w Levant_N i A – wzór, który może powstać tylko wtedy, gdy Levant_ChL jest produktem domieszki między grupami spokrewnionymi, być może daleko, z Levant_N i A35. Najbardziej negatywne statystyki f3- i f4 powstają, gdy A jest populacją z Iranu lub Kaukazu. Sugeruje to, że populacja Levant_ChL wywodzi się z populacji spokrewnionej z Levant_N, ale posiada również przodków z nielewantyńskich populacji spokrewnionych z tymi z Iranu lub Kaukazu, z którymi Levant_N nie dzieli się (lub przynajmniej dzieli się w takim samym stopniu).

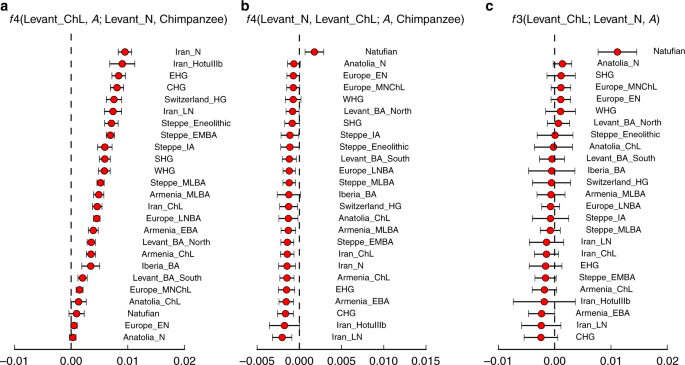
Charakterystyka genetyczna Levant_ChL. a Statystyka f4(Levant_ChL, A; Levant_N, Chimpanzee) pokazuje bliskie pokrewieństwo między neolitycznymi i chalkolitycznymi populacjami Lewantu, ponieważ neolit lewantyński dzieli więcej alleli z chalkolitycznym Lewantem niż z jakąkolwiek inną populacją. b Statystyka f4(Levant_N, Levant_ChL; A, szympans) pokazuje asymetryczną relację między Levant_N i Levant_ChL a innymi starożytnymi populacjami zachodnioeuropejskimi. Statystyka jest najbardziej ujemna dla populacji z Iranu i Kaukazu, wskazując, że Levant_ChL dzieli z nimi więcej alleli niż Levant_N. c Statystyka f3(Levant_ChL; Levant_N, A) testuje sygnały domieszki w Levant_ChL. Ujemna statystyka f3 wskazuje, że populacja Levant_ChL jest domieszkowana. Populacje z Iranu i Kaukazu dają najwięcej ujemnych statystyk. Szacowana statystyka ± 3 błędy standardowe jest wskazana
Przodkowie ludu chalkolitu z Lewantu
Użyliśmy qpAdm jako naszego głównego narzędzia do identyfikacji wiarygodnych modeli domieszek dla starożytnych populacji, dla których mamy dane (więcej informacji znajduje się w uwadze dodatkowej 3 ) 36 .
Metoda qpAdm ocenia, czy badany zbiór N „lewych” populacji — w tym populacja „docelowa” (populacja, której pochodzenie jest modelowane) oraz zbiór N − 1 dodatkowych populacji — jest zgodny z otrzymanymi z mieszanin w różnych proporcjach z N - 1 populacji przodków spokrewnionych w różny sposób ze zbiorem populacji obcych, określanych jako populacje „prawe”. We wszystkich naszych analizach używamy podstawowego zestawu 11 „prawicowych” grup zewnętrznych określanych zbiorczo jako „09NW” – Ust_Ishim, Kostenki14, MA1, Han, Papuan, Onge, Czukczi, Karitiana, Mbuti, Natufian i WHG – których wartość dla rozplątywanie rozbieżnych szczepów przodków obecnych u starożytnych mieszkańców Bliskiego Wschodu zostało udokumentowane w Lazaridis i in. 24(dla niektórych analiz uzupełniamy ten zbiór o dodatkowe grupy obce). Aby ocenić, czy populacje „lewe” są zgodne z hipotezą wyprowadzenia z N − 1 źródeł, qpAdm efektywnie oblicza wszystkie możliwe statystyki postaci f 4 (Left i , Left j ; Right k , Right l ), dla wszystkich możliwych pary populacji w proponowanych zbiorach „Lewy” i „Prawy ” . Następnie określa, czy wszystkie statystyki można zapisać jako liniową kombinację statystyk f 4 odpowiadających wzorcom różnicowania między proponowanymi N − 1 populacji przodków, odpowiednio uwzględniając kowariancję tych statystyk i obliczając pojedynczą wartość p dla dopasowania na podstawie rozkładu T -kwadrat Hotellinga 36 . Dla modeli, które są zgodne z danymi ( p > 0,05), qpAdm szacuje proporcje domieszki dla populacji docelowej ze źródeł związanych z populacjami przodków N − 1 (z błędami standardowymi). Co najważniejsze, qpAdm nie wymaga określania jawnego modelu, w jaki sposób populacje „prawej” grupy zewnętrznej są powiązane.
Najpierw zbadaliśmy wszystkie możliwe zestawy populacji „lewicowych”, które składały się z Levant_ChL wraz z jedną inną starożytną populacją ze zbioru danych analizy. Testując szeroki zakres starożytnych populacji, stwierdziliśmy, że wartości p dla wszystkich możliwych populacji lewicowych były poniżej 0, 05 (dane uzupełniające 2), pokazując, że Levant_ChL nie jest zgodny z byciem kladem z którymkolwiek z nich w stosunku do „prawej” grupy zewnętrznej 09NW. Następnie rozważyliśmy modele z „lewicowymi” zestawami populacji zawierającymi Levant_ChL wraz z dwiema dodatkowymi starożytnymi populacjami, co odpowiada modelowaniu Levant_ChL w wyniku dwukierunkowej domieszki między populacjami związanymi z tymi dwoma innymi starożytnymi populacjami. Aby zmniejszyć liczbę testowanych hipotez, ograniczyliśmy modele do par populacji źródłowych, które zawierają co najmniej jedną z sześciu populacji, które uważamy za najbardziej prawdopodobne źródła domieszek w oparciu o bliskość geograficzną i czasową: Anatolia_N, Anatolia_ChL, Armenia_ChL, Iran_ChL , Iran_N i Lewant_N. Ponownie, nie znajdujemy wiarygodnych dwukierunkowych modeli domieszek przy użyciu progu p > 0, 05 (rysunek uzupełniający 2 i dane uzupełniające 3 ). Na koniec przetestowaliśmy możliwe trójdrożne zdarzenia domieszkowe, ograniczając się do trojaczków, które zawierają co najmniej dwa z sześciu najbardziej prawdopodobnych źródeł domieszek. Prawdopodobne rozwiązania przy p > 0, 05 wymieniono w Tabeli 1 (pełne wyniki przedstawiono na Rycinie uzupełniającej 3 i Danych uzupełniających 4 ).
Znaleźliśmy wielu kandydatów do trójdrożnych modeli domieszek, zawsze obejmujących (1) Levant_N (2) Anatolia_N lub Europe_EN oraz (3) Iran_ChL, Iran_N, Iran_LN, Iran_HotuIIIb lub Levant_BA_North. Są to wszystkie bardzo podobne modele, ponieważ Europe_EN (pierwsi europejscy rolnicy) są genetycznie wywodzący się głównie od anatolijskich rolników (Anatolia_N) 31 , a Levant_BA_North ma przodków spokrewnionych z Levant_N i Iran_ChL 26. Aby rozróżnić modele obejmujące anatolijski neolit (Anatolia_N) i europejski wczesny neolit (Europe_EN), powtórzyliśmy analizę, włączając dodatkowe populacje z grupy zewnętrznej w zestawie „Prawy”, które są wrażliwe na europejską domieszkę związaną z łowcami-zbieraczami obecną w większym stopniu w Europe_EN niż w Anatolia_N (rysunek uzupełniający 4a ) 31 (w ten sposób dodaliśmy Switzerland_HG, SHG, EHG, Iberia_BA, Steppe_Eneolithic, Europe_MNChL, Europe_LNBA do „prawych” grup zewnętrznych; skróty w tabeli uzupełniającej 2 ). Stwierdziliśmy, że tylko modele obejmujące Levant_N, Anatolia_N i Iran_ChL lub Levant_BA_North przeszły przy p > 0, 05 (Tabela 1). Aby rozróżnić Iran_ChL i Levant_BA_North, dodaliśmy Iran_N do zestawu grup zewnętrznych (w sumie 19 = 11 + 8 grup zewnętrznych) (Rysunek dodatkowy 4b ). Tylko model z udziałem Iran_ChL pozostał wiarygodny. Na podstawie tego wyjątkowo dopasowanego modelu qpAdm wnioskujemy, że pochodzenie Levant_ChL jest wynikiem trójstronnej domieszki populacji związanych z Levant_N (57%), Iran_ChL (17%) i Anatolia_N (26%).
Pochodzenie populacji późnej lewantyńskiej epoki brązu
Uderzyło nas, że wcześniej opublikowane próbki lewantyńskie z epoki brązu z miejsc „Ain Ghazal w dzisiejszej Jordanii (Levant_BA_South) i Sidon w dzisiejszym Libanie (Levant_BA_North) można modelować jako domieszki dwukierunkowe, bez wkładu Anatolia_N co jest wymagane do modelowania populacji Levant_ChL 24 , 26 . Sugeruje to, że populacja Levant_ChL może nie być bezpośrednio przodkiem tych późniejszych populacji lewantyńskich z epoki brązu, ponieważ gdyby tak było, spodziewalibyśmy się również wykrycia składnika pochodzenia Anatolia_N. Poniżej traktujemy Levant_BA_South i Levant_BA_North jako oddzielne populacje do analizy, ponieważ statystyka symetrii f 4(Levant_BA_North, Levant_BA_South; A, szympans) jest istotny dla liczby populacji testowych A (| Z | ≥ 3) (dane uzupełniające 5 ), zgodnie z różnymi szacunkowymi proporcjami przodków Levant_N i Iran_ChL zgłoszonymi w 24 , 26 .
Aby przetestować hipotezę, że Levant_ChL może być bezpośrednio przodkiem populacji lewantyńskiej epoki brązu, próbowaliśmy modelować zarówno Levant_BA_South, jak i Levant_BA_North jako dwukierunkowe domieszki między Levant_ChL i każdą inną starożytną populacją w naszym zbiorze danych, używając podstawowego zestawu populacji 09NW jako „Prawe” grupy zewnętrzne. Porównaliśmy również te modele z wcześniej opublikowanymi modelami, które wykorzystywały populacje Levant_N i Iran_ChL jako źródła (Tabela 2 ; Rysunek uzupełniający 5 ; Dane uzupełniające 6). W przypadku Levant_BA_South z Ain Ghazal w Jordanii wiele modeli było wiarygodnych, dlatego powróciliśmy do strategii dodawania dodatkowych „prawicowych” grup zewnętrznych, które są różnie spokrewnione z jedną lub kilkoma populacjami „lewicowymi” (konkretnie my dodano różne kombinacje Armenia_EBA, Steppe_EMBA, Switzerland_HG, Iran_LN i Iran_N). Tylko model obejmujący Levant_N i Iran_ChL pozostaje wiarygodny we wszystkich warunkach. Możemy zatem stwierdzić, że grupy związane z Levant_ChL wniosły niewielki wkład w pochodzenie Levant_BA_South.
Obserwujemy jakościowo odmienny wzorzec w próbkach Levant_BA_North z Sydonu w Libanie, gdzie modele obejmujące Levant_ChL w połączeniu z populacjami Iran_N, Iran_LN lub Iran_HotuIIIb wydają się być znacznie lepiej dopasowane niż modele obejmujące Levant_N + Iran_ChL. W dużej mierze potwierdzamy ten wynik, stosując „prawe” grupy zewnętrzne populacji zdefiniowane w Haber et al. 26 (skr. Haber: Ust_Ishim, Kostenki14, MA1, Han, Papuan, Ami, Chuckhi, Karitiana, Mbuti, Switzerland_HG, EHG, WHG i CHG), chociaż stwierdzamy, że konkretny model obejmujący Iran_HotuIIIb nie działa już z tym „Prawym ” zbiór populacji. Badając to dalej, stwierdzamy, że dodanie Anatolia_N do zestawu „prawej” grupy zewnętrznej wyklucza model Levant_N + Iran_ChL preferowany przez Habera i in. 26. Wyniki te sugerują, że populacja, która miała przodków bliżej spokrewnionych z Levant_ChL niż z Levant_N, przyczyniła się do populacji Levant_BA_North, nawet jeśli nie przyczyniła się w sposób wykrywalny do populacji Levant_BA_South.
Uzyskaliśmy dodatkowy wgląd, uruchamiając qpAdm z Levant_BA_South jako cel dwukierunkowej domieszki między Levant_N i Iran_ChL, ale teraz dodając Levant_ChL i Anatolia_N do podstawowego zestawu 09NW „Right ” 11 grup zewnętrznych. Dodanie Levant_ChL powoduje niepowodzenie modelu, co wskazuje, że Levant_BA_South i Levant_ChL mają wspólne pochodzenie po oddzieleniu ich obu od przodków Levant_N i Iran_ChL. Tak więc w przeszłości istniała niespróbowana populacja, która przyczyniła się zarówno do Levant_ChL, jak i do Levant_BA_South, mimo że Levant_ChL nie może być bezpośrednim przodkiem Levant_BA_South, ponieważ, jak opisano powyżej, ma pochodzenie związane z Anatolią_N, którego nie ma w Levant_BA_South.
Heterogeniczność genetyczna w lewantyńskiej epoce brązu
Obawialiśmy się, że nasze odkrycie, że populacja Levant_ChL była mieszanką co najmniej trzech grup, może być artefaktem braku dostępu do próbek blisko spokrewnionych z prawdziwymi populacjami przodków. Jedną z konkretnych możliwości, które rozważaliśmy, jest to, że pojedyncza populacja przodków została zmieszana z Lewantem, aby przyczynić się zarówno do populacji Levant_ChL, jak i Levant_BA_South, i że była to populacja bez próby na klinie domieszek między Anatolią_N i Iran_ChL, wyjaśniając, dlaczego qpAdm wymaga trzech populacji źródłowych do wymodeluj to. Aby formalnie przetestować tę hipotezę, użyliśmy qpWave 36 , 37 , 38 , który określa minimalną liczbę populacji źródłowych wymaganych do modelowania relacji między „ ”populacjami lewicy w stosunku do”” populacje pozagrupowe. W przeciwieństwie do qpAdm, qpWave nie wymaga, aby do analizy były dostępne populacje blisko spokrewnione z prawdziwymi populacjami źródłowymi. Zamiast tego traktuje jednakowo wszystkie „lewicowe” populacje i próbuje określić minimalną liczbę teoretycznych populacji źródłowych wymaganych do modelowania zbioru „lewicowych” populacji w stosunku do zewnętrznych grup populacji „prawicowych”. Dlatego modelujemy związek między Levant_N, Levant_ChL i Levant_BA_South jako populacje „lewe ” w stosunku do populacji „prawej” grupy zewnętrznej 09NW (Tabela 3). Stwierdzamy, że do modelowania pochodzenia tych populacji lewantyńskich nadal wymagane są co najmniej trzy populacje źródłowe, wspierając model, w którym co najmniej trzy oddzielne źródła przodków są obecne w Lewancie między neolitem, chalkolitem i epoką brązu.
Ponownie zastosowaliśmy qpWave, zastępując Levant_ChL Levant_BA_North i stwierdziliśmy, że minimalna liczba populacji źródłowych to tylko dwie. Jednak gdy uwzględnimy populację Levant_ChL jako dodatkową grupę zewnętrzną, ponownie wymagane są trzy populacje źródłowe. Sugeruje to, że przy braku danych z Levant_ChL nie ma wystarczającej dźwigni statystycznej do wykrycia pochodzenia związanego z Anatolią, które jest naprawdę obecne w postaci zmieszanej w populacji Levant_BA_North (dane z populacji Levant_ChL umożliwiają wykrycie tego przodka). To może wyjaśniać, dlaczego Haber i in. 26 nie wykrył domieszki związanej z anatolijskim neolitem w Levant_BA_North.
Biologicznie ważne mutacje w populacji Peqi’in
To badanie prawie podwaja liczbę osobników z danymi całego genomu ze starożytnego Lewantu. Mierzony w kategoriach średniego zasięgu w SNP, wzrost ten jest jeszcze wyraźniejszy ze względu na wyższą jakość przedstawionych tutaj danych niż w poprzednich badaniach starożytnych mieszkańców Bliskiego Wschodu 24 , 26 . Zatem niniejsze badanie znacznie zwiększa moc analizy zmiany częstotliwości alleli, o których wiadomo, że są ważne biologicznie.
Wykorzystaliśmy nasze dane do zbadania zmiany częstości alleli SNP, o których wiadomo, że są związane z metabolizmem, pigmentacją, podatnością na choroby, odpornością i stanem zapalnym w populacji Levant_ChL, rozważanej w odniesieniu do częstości alleli w Levant_N, Levant_BA_North, Levant_BA_South, Anatolia_N i Populacje Iran_ChL i obecne pule pochodzenia afrykańskiego (AFR), wschodnioazjatyckiego (EAS) i europejskiego (EUR) w zbiorze danych fazy 3 projektu 1000 genomów 39 (dane uzupełniające 7 ).
Zwracamy uwagę na trzy interesujące ustalenia. Po pierwsze, allel (G) w rs12913832 w pobliżu genu OCA2, z udowodnionym powiązaniem z niebieskim kolorem oczu u osób pochodzenia europejskiego40 , ma szacunkową częstość alleli alternatywnych wynosząca 49% w populacji Levant_ChL, co sugeruje, że fenotyp niebieskookich był powszechny w Levant_ChL.
Po drugie, allel w rs1426654 w genie SLC24A5 , który jest jednym z najważniejszych wyznaczników jasnej pigmentacji u mieszkańców Zachodniej Eurazji41, jest ustalony dla pochodnego allelu ( A ) w populacji Levant_ChL, co sugeruje, że fenotyp o jasnej karnacji mógł być powszechny w tej populacji, chociaż wszelkie wnioski dotyczące pigmentacji skóry na podstawie częstości alleli obserwowanych w jednym miejscu należy traktować z ostrożnością 42 .
Po trzecie, allel ( G ) w rs6903823 w genach ZKSCAN3 i ZSCAN31 , który jest nieobecny u wszystkich wczesnych rolników zgłoszonych do tej pory (Levant_N, Anatolia_N, Iran_N) i który, jak argumentowano, był pod pozytywną selekcją przez Mathieson i in. 31 , występuje z szacowaną częstością 20% w populacjach Levant_ChL, 17% w Levant_BA_South i 15% w populacjach Iran_ChL, podczas gdy nie występuje we wszystkich innych populacjach. Sugeruje to, że częstość allelu rosła w populacjach z epoki chalkolitu i epoki brązu na Bliskim Wschodzie w tym samym czasie, gdy rosła w Europie.
Dyskusja
Okres chalkolitu w Lewancie był świadkiem poważnych przemian kulturowych praktycznie we wszystkich obszarach kultury, w tym w produkcji rzemieślniczej, praktykach pogrzebowych i rytualnych, wzorcach osadnictwa oraz ekspresji ikonograficznej i symbolicznej 43. Obecne badanie zapewnia wgląd w długotrwałą debatę w prehistorii Lewantu, sugerując, że pojawienie się chalkolitycznej kultury materialnej było związane z ruchem i obrotem populacji.
Jakość starożytnego DNA uzyskanego z próbek jaskini Peqi’in jest doskonała w porównaniu z innymi miejscami na Bliskim Wschodzie. Stawiamy hipotezę, że wyjątkowe zachowanie wynika z dwóch czynników. Po pierwsze, ukierunkowane pobieranie próbek starożytnego DNA z części skalistej kości skroniowej umożliwia uzyskanie wysokiej jakości starożytnego DNA z wcześniej niedostępnych regionów geograficznych 24 , 27 , 44 , 45 . Po drugie, środowisko jaskini Peqi’in prawdopodobnie sprzyja zachowaniu DNA. Szczątki szkieletu — albo przechowywane w ossuariach, albo złożone w ziemi — zostały szybko pokryte wapienną skorupą, izolując je od bezpośredniego otoczenia i chroniąc przed kwaśnymi warunkami, o których wiadomo, że uszkadzają DNA.
Odkryliśmy, że osoby pochowane w jaskini Peqi’in reprezentują stosunkowo jednorodną genetycznie populację. Ta jednorodność jest widoczna nie tylko w analizach całego genomu, ale także w fakcie, że większość osobników płci męskiej (dziewięć na dziesięć) należy do haplogrupy T chromosomu Y (patrz tabela uzupełniająca 1), linii, o której sądzono, że zróżnicowana na Bliskim Wschodzie 46 . Odkrycie to kontrastuje zarówno z wcześniejszymi (neolitycznymi i epipaleolitycznymi) populacjami lewantyńskimi, które były zdominowane przez haplogrupę E 24, jak i osobnikami z późniejszej epoki brązu, z których wszystkie należały do haplogrupy J 24 , 26 .
Nasze odkrycie, że populację Levant_ChL można dobrze modelować jako trójskładnikową domieszkę między Levant_N (57%), Anatolią_N (26%) i Iran_ChL (17%), podczas gdy Levant_BA_South można modelować jako mieszaninę Levant_N (58 %) i Iran_ChL (42%), ale ma niewiele, jeśli w ogóle, dodatkowych przodków związanych z Anatolią_N, można wytłumaczyć jedynie wieloma epizodami przemieszczania się populacji. Obecność przodków spokrewnionych z Iranem w obu populacjach – ale nie we wcześniejszym Levant_N – sugeruje historię rozprzestrzeniania się do Lewantu ludów spokrewnionych z irańskimi rolnikami, co musiało nastąpić co najmniej do czasów chalkolitu. Składnik Anatolian_N obecny w próbce Levant_ChL, ale nie w próbce Levant_BA_South sugeruje, że istniało również oddzielne rozprzestrzenianie się ludzi spokrewnionych z Anatolią w regionie. Populacja Levant_BA_South może zatem reprezentować pozostałość populacji, która powstała po początkowym rozprzestrzenieniu się pochodzenia związanego z Iran_ChL do Lewantu, na który nie miało wpływu rozprzestrzenianie się populacji związanej z Anatolią_N, lub być może ponowne wprowadzenie populacji bez populacji związanej z Anatolią_N przodków do regionu. Ponadto stwierdzamy, że populacja Levant_ChL nie służy jako prawdopodobne źródło przodków związanych z Lewantyną we współczesnych populacjach Afryki Wschodniej (patrz Uwaga dodatkowa 4 ) 24 .
Te wyniki genetyczne mają uderzające korelaty ze zmianami kultury materialnej w zapisie archeologicznym. Znaleziska archeologiczne w jaskini Peqi’in mają charakterystyczne cechy charakterystyczne dla innych stanowisk chalkolitycznych, zarówno na północy, jak i na południu, w tym wtórne pochówki w ossuariach z ikonograficznymi i geometrycznymi wzorami. Sugerowano, że niektóre zwyczaje, artefakty i motywy pogrzebowe z późnego chalkolitu mogły mieć swój początek we wcześniejszych tradycjach neolitycznych w Anatolii i północnej Mezopotamii 8 , 13 , 47 . Niektóre z form ekspresji artystycznej były związane ze znaleziskami i ideami oraz z późniejszymi koncepcjami religijnymi, takimi jak bogowie Inanna i Dumuzi z tych bardziej północnych regionów 6 , 8 , 47 ,48, 49 , 50 . Postawiono hipotezę, że wiedza i zasoby potrzebne do wytworzenia artefaktów metalurgicznych w Lewancie pochodzą z północy 11,51 .
Nasze odkrycie nieciągłości genetycznej między okresami chalkolitu i wczesnej epoki brązu rezonuje również z aspektami zapisów archeologicznych naznaczonych dramatycznymi zmianami we wzorcach osadnictwa 43 , porzucaniem stanowisk na dużą skalę 52 , 53 , 54 , 55 , znacznie mniejszą liczbą przedmiotów o znaczeniu symbolicznym oraz zmiany w praktykach pochówku, w tym zniknięcie wtórnego pochówku w ossuariach 56 , 57 , 58 , 59 . Potwierdza to pogląd, że głęboki przewrót kulturowy, prowadzący do wyginięcia populacji, wiązał się z upadkiem kultury chalkolitu w tym regionie 18 , 60, 61 , 62 , 63 , 64 .
Te starożytne wyniki DNA ujawniają stosunkowo jednorodną genetycznie populację w Peqi’in. Pokazujemy, że ruchy ludności w regionie południowego Lewantu były niezwykle dynamiczne, a niektóre populacje, takie jak ta pochowana w Peqi’in, zostały częściowo ukształtowane przez wpływy egzogeniczne. Badanie to zawiera również studium przypadku istotne poza Lewantem, pokazujące, w jaki sposób połączona analiza danych genetycznych i archeologicznych może dostarczyć bogatych informacji na temat mechanizmu zmian w przeszłych społeczeństwach.
Metody
Generowanie danych
Jaskinia grobowa Peqi’in została wykopana pod auspicjami Izraelskiego Urzędu Starożytności (zezwolenie nr 2297/1995). Przeszukaliśmy 46 elementów ludzkiego szkieletu z jaskini Peqi’in, z których 37 to kamienne fragmenty kości skroniowej. Przygotowaliśmy od 15 do 114 mg proszku kostnego dla każdej próbki, wiercąc ze zbitej części próbki po oczyszczeniu powierzchni narzędziem Dremel lub wiercąc w wewnętrznej części ucha skalistej części kości skroniowej 27 . Ekstrahowaliśmy DNA przy użyciu protokołu ekstrakcji opartego na kolumnie krzemionkowej, który został zoptymalizowany pod kątem ekstrakcji starożytnego DNA 28 , modyfikując protokoły, zastępując zespół kolumny MinElute wstępnie zmontowanym urządzeniem z kolumną wirującą, jak w Korlević i in. 65. Do proszku kostnego dodaliśmy 1,5 ml buforu ekstrakcyjnego (0,45 M EDTA, pH 8,0 (BioExpress), 0,05% proteinazy K (Sigma)) i inkubowaliśmy w 2,0 ml probówkach w temperaturze 37°C przez noc, obracając. Po inkubacji wirowaliśmy próbki z maksymalną prędkością przez 2 minuty i dodaliśmy 13 ml buforu wiążącego (5 M GuHCl (Sigma), 40% izopropanolu (Sigma), 400 μg octanu sodu (Sigma), pH 5, 2 (Sigma)) do supernatantu. Przenieśliśmy mieszaninę do High Pure Extender z zestawu Viral Nucleic Acid Large Volume Kit (Roche) i wirowaliśmy przy 2000 x g , aż cały płyn zniknął z lejka. Odłączyliśmy kolumnę krzemionkową od lejka, umieściliśmy ją w świeżej 2 ml probówce zbierającej i wirowaliśmy przez 1 minutę przy 8000 x g. Przeprowadziliśmy dwa przemycia, dodając 700 μl buforu PE (Qiagen) do kolumn i wirowano przy 8000 x g przez 30 s, wymieniając probówkę zbierającą po każdym przemyciu. Wykonaliśmy wirowanie na sucho z maksymalną prędkością przez 1 minutę, a następnie wymieniliśmy probówkę zbiorczą. Usunęliśmy eluat DNA z kolumny, dodając 45 μl TTE (10 mM Tris-HCl, pH 8,0 (ThermoFisher), 1 mM EDTA, pH 9,0 (BioExpress), 0,05% Tween-20 (Sigma)) do matrycy krzemionkowej , inkubując przez 5 minut, a następnie wirowano z maksymalną prędkością przez 1 minutę. Powtarzaliśmy ten krok, aż uzyskaliśmy całkowitą objętość 90 μl. W przypadku ponownych prób jednej z próbek przemyliśmy proszek 1 ml 0,5% wybielacza (inkubacja przez 15 minut), a następnie trzykrotnie przemyto 1 ml wody (inkubacja 3 minuty), przed ekstrakcją DNA, jak opisano w Korlević i in. 65 (patrz Dane uzupełniające 1 ) i przygotowane biblioteki przy użyciu częściowego leczenia UDG 29(protokoły biblioteczne różniły się nieznacznie w trakcie generowania danych, patrz Dane uzupełniające 1). Dodaliśmy 30 μl ekstraktu do mieszaniny do traktowania USER (1× Buffer Tango (ThermoFisher), 100 μM dNTP Mix (ThermoFisher), 1 mM ATP (ThermoFisher), 0,06 U/μL enzymu USER (NEB)) i inkubowaliśmy reakcję w 37°C przez 30 min. Zahamowaliśmy enzym UDG przez dodanie inhibitora glikozylazy uracylu (0,12 U / μl; NEB) do mieszanki i inkubację przez kolejne 30 minut w 37 ° C. Następnie wykonaliśmy naprawę tępych końców na próbkach, dodając T4 PNK (0,5 U / μl; ThermoFisher) i polimerazę T4 (90,1 U / μl; ThermoFisher) do mieszaniny i inkubując przez 15 min w 25 ° C, a następnie przez 5 min w temp. 12°C. Oczyściliśmy reakcje za pomocą zestawu do oczyszczania MinElute PCR, dodając pięć objętości buforu PB do mieszaniny reakcyjnej, przenosząc do probówki zbiorczej i wirując przez 30 s przy 3300 × g. Odrzuciliśmy płyn i przemyliśmy dwukrotnie, dodając 700 μl buforu PE do kolumny, wirując przez 30 s przy maksymalnej prędkości i odrzucając probówkę zbierającą, a następnie wirując na sucho przez 1 minutę przy maksymalnej prędkości. Eluowaliśmy próbki w 18 μl 10 mM Tris-HCl (ThermoFisher), który dodaliśmy do membrany krzemionkowej i pozostawiliśmy na 5 minut, a następnie wirowano przez 1 minutę z maksymalną prędkością. Połączyliśmy unikalne adaptery z cząsteczkami w każdej próbce, inkubując mieszaninę próbek w mieszaninie reakcyjnej do ligacji (1 x bufor do ligazy DNA T4 (ThermoFisher), 5% PEG-4000 (ThermoFisher), 0,25 μM adapter P5 (patrz ref. 29 sugerowane informacje dotyczące przygotowania), adapter 0,25 μM P7 (patrz ref. 29sugerowane informacje dotyczące przygotowania), 0,125 U/μl ligazy DNA T4 (ThermoFisher)) przez 30 minut w temperaturze pokojowej. Oczyściliśmy mieszaninę ligacyjną, stosując procedurę oczyszczania opisaną powyżej, eluując 20 μl 10 mM Tris-HCl. Wypełniliśmy zligowane adaptery, dodając mieszaninę reakcyjną do napełniania (1 × bufor ThermoPol (NEB), 250 μM dNTP Mix (ThermoFisher), 0,4 U / μL polimerazy Bst, duży fragment (NEB)) do produktu ligacji i inkubacja w 37°C przez 20 min, a następnie w 80°C przez 20 min. Na koniec amplifikowaliśmy biblioteki za pomocą PCR, dodając 39 μl produktu reakcji wypełnienia do mieszaniny reakcyjnej PCR (1 x Pfu Turbo Cx Reaction Buffer (Agilent Technologies), 0, 4 μM PreHyb-F (5′-CTTTCCCTACACGACGCTTC-3′ ), 0,4 μM PreHyb-R (5′-GTGACTGGAGTTCAGACGTGTGCT-3′), 0,2 mM dNTP Mix (ThermoFisher), Polimeraza DNA 5U Pfu Turbo Cx Hotstart (Agilent Technologies)). Podzieliliśmy każdą reakcję na porcje po 50 μl i umieściliśmy w szczelnie zamkniętej płytce do PCR. Przeprowadziliśmy wstępną denaturację próbek przez 2 minuty w 95 ° C, a następnie 30 cykli w 95 ° C przez 30 s, 55 ° C przez 30 s, 72 ° C przez 1 minutę i wykonaliśmy końcowe wydłużanie w 72 ° C przez 10 min.
Przeszukaliśmy wszystkie biblioteki pod kątem autentycznego DNA poprzez wzbogacenie genomu mitochondrialnego i 50 docelowych loci jądrowych, a następnie sekwencjonowanie na instrumencie Illumina NextSeq500 przez 2 × 75 cykli i 2 × 7 cykli w celu odczytania wskaźników. Wzbogaciliśmy obiecujące biblioteki o około 1, 2 M SNP, jak opisano w ref. 31 , 36 , 66 ,]a następnie zsekwencjonowano na sekwenatorze NextSeq500 stosując sekwencje sparowanych końców o długości 75 par zasad. Podczas przetwarzania obliczeniowego początkowo usunęliśmy identyfikujące sekwencje oligonukleotydowe i adaptery, oddzielając poszczególne próbki od połączonych przechwyceń przez ich identyfikujące 7-parowe indeksy na końcach 5 'i 3′ odczytów i wymagając dopasowań do kodów kreskowych specyficznych dla próbki dołączonych bezpośrednio do sekwencji fragmentów, dopuszczając nie więcej niż jedną niezgodność na indeks/kod kreskowy. Użyliśmy SeqPrep 67 do usunięcia adapterów, a także do łączenia sparowanych odczytów końcowych w pojedyncze sekwencje, wymagając nakładania się co najmniej 15 par zasad (pozwalając na jedno niedopasowanie), używając bazy najwyższej jakości w połączonym regionie, w którym wystąpił konflikt. Użyliśmy samse w bwa (v0.6.1) 68wyrównać odczyty. W eksperymencie wzbogacania mitochondrialnego DNA dostosowaliśmy się do genomu mitochondrialnego RSRS 69 . W eksperymencie wzbogacania całego genomu dopasowaliśmy się do genomu referencyjnego hg19. Zidentyfikowaliśmy zduplikowane sekwencje jako te z tymi samymi pozycjami początkowymi i końcowymi oraz orientacją, a także identycznymi parami kodów kreskowych i zachowaliśmy sekwencję najwyższej jakości z każdego duplikatu. Wykonaliśmy pseudo-haploidalne wywołania SNP dla każdej pozycji, używając losowo wybranej sekwencji obejmującej każde miejsce docelowe, usuwając dwie zasady na końcach każdej sekwencji, aby usunąć mutacje deaminowane i wymagając minimalnej jakości mapowania (MAPQ ≥ 10) i ograniczając się do strony o minimalnej podstawowej jakości (≥20).
Oceniliśmy jakość każdej biblioteki na etapie badań przesiewowych, stosując trzy standardowe metody określania autentyczności starożytnego DNA. Najpierw przeanalizowaliśmy dane genomu mitochondrialnego, aby określić stopień dopasowania do sekwencji konsensusowej, używając contamMix 30 . Po drugie, ograniczyliśmy się do próbek, w których wskaźnik podstawień C-to-T w końcowych nukleotydach wynosił co najmniej 3%, zgodnie z oczekiwaniami dla prawdziwego starożytnego DNA przy użyciu protokołu częściowego leczenia UDG 29. Na koniec wykorzystaliśmy oprogramowanie ANGSD, aby uzyskać konserwatywne oszacowanie zanieczyszczenia chromosomu X osób określonych jako męskie w oparciu o szybkość polimorfizmu sekwencji chromosomu X (mężczyźni mają tylko jeden chromosom X, więc nie oczekuje się wykazać polimorfizm); wykluczyliśmy biblioteki z szacunkami zanieczyszczenia X większymi niż 1,5% 32 . W przypadku próbek, w których wyprodukowano wiele bibliotek dla jednego osobnika, połączyliśmy biblioteki, które przeszły kontrolę jakości i uzyskaliśmy nowe pseudo-haploidalne wywołania SNP.
Określiliśmy haplogrupy mitochondrialnego DNA za pomocą narzędzia haplogrep2 70 , stosując sekwencję konsensusową zbudowaną z odczytów wzbogaconych o genom mitochondrialny, ograniczając się do uszkodzonych odczytów przy użyciu PMDtools 71 (pmdscore ≥ 3) i przycinając 5 zasad z każdego końca, aby znacznie zmniejszyć wskaźnik błędów w wyniku deaminacji.
Starożytne DNA stanowi wyzwanie przy przypisywaniu haplogrup chromosomu Y ze względu na możliwość zanieczyszczenia, uszkodzenia DNA lub brakujących danych. Aby przypisać haplogrupy Y do naszych danych, wykorzystaliśmy zmodyfikowaną wersję procedury stosowanej w analizie współczesnych chromosomów Y w projekcie 1000 genomów 72, który wykorzystuje przeszukiwanie wszerz w celu przeszukania drzewa chromosomu Y. Wezwania wykonaliśmy na drzewie ISOGG od 04.01.2016 [ http://isogg.org] i zmodyfikował wywołującego, aby wyprowadzał wywołania alleli pochodnych i przodków dla każdej pozycji informacyjnej w drzewie. Następnie przypisaliśmy wynik do każdej z haplogrup referencyjnych, licząc liczbę niedopasowań w liczbie obserwowanych alleli pochodnych na tej gałęzi i zmniejszonych mutacjach pochodnych, które były przejściami do 1/3 transwersji, aby uwzględnić błędy związane z uszkodzeniem DNA . Przypisaliśmy próbkę do haplogrupy referencyjnej z najbliższym dopasowaniem na podstawie tego wyniku. Chociaż staraliśmy się wywołać wezwanie dla każdej próbki, zauważamy, że próbki z mniej niż 100 000 SNP mają zbyt mało danych, aby z pewnością zidentyfikować prawidłową haplogrupę, i zachęcamy do ostrożności przy interpretacji tych wyników.
Dane z 22 próbek, które przeszły testy zanieczyszczenia i kontroli jakości, przedstawiono w tabeli uzupełniającej 1 , przy średnim pokryciu 0, 97 × na 1240 k celów SNP i średnio 358 313 SNP objętych co najmniej raz. Tabela według biblioteki opisująca wyniki badań przesiewowych jest przedstawiona w danych uzupełniających 1 . Wykluczyliśmy dwie osoby z dalszej analizy, ponieważ wzorce genetyczne zaobserwowane przy użyciu metody opisanej w Kuhn et al. 73 . Pokazano, że byli krewnymi pierwszego stopnia próbek o wyższym pokryciu w zbiorze danych. Ograniczyliśmy dane z próbki I1183, aby obejmowały tylko sekwencje z dowodami podstawienia C-to-T, aby zminimalizować zanieczyszczenie, które było widoczne w pełnych danych z tych próbek.
Połączyliśmy nowo zgłoszone dane z istniejącymi danymi z Lazaridis i in. 24 oraz Haber i in. 26 , używając programu scalania firmy EIGENSOFT 33 . Powstałe zestawy danych, określane jako HO + i HOIll+, zawierają 20 nowych niepowiązanych próbek połączonych z HO i HOI ll z Lazaridis i in. 24 i 5 starożytnych próbek z Sydonu w Libanie (nazwa populacji: Levant_BA_North) z Haber et al. 26 , odpowiednio. HO+ obejmuje dane od 2891 współczesnych i starożytnych osobników z 591 642 SNP, a HOIll+ zawiera dane od 306 starożytnych osobników z 1 054 637 SNP.
Analiza głównych składowych
Przeprowadziliśmy PCA na zbiorze danych HO + przy użyciu smartpca 33 . Wykorzystaliśmy w sumie 984 współczesnych osobników do PCA i przewidzieliśmy 306 starożytnych próbek. Użyliśmy domyślnych parametrów z ustawieniami lsqproject: YES i numoutlieriter: 0. Oszacowaliśmy F ST za pomocą smartpca dla 21 starożytnych populacji Bliskiego Wschodu składających się z więcej niż jednego osobnika i 8 współczesnych populacji przy użyciu parametrów domyślnych, z chowem wsobnym: TAK i fstonly: TAK (Rysunek dodatkowy 1 ) . Przeprowadziliśmy analizy przy użyciu zestawu danych HO +.
Domieszka
Przeprowadziliśmy analizę ADMIXTURE 34 na zbiorze danych HO+. Przed analizami przycinaliśmy SNP w silnej nierównowadze powiązań ze sobą za pomocą PLINK 74 przy użyciu parametrów — niezależnie od par 200 25 0,4. Przeprowadziliśmy analizę ADMIXTURE na 3 00 885 SNP pozostałych w przyciętym zbiorze danych. Dla każdej wartości k między 2 a 14 wykonaliśmy 20 analiz powtórzeń i zachowaliśmy replikację o najwyższym prawdopodobieństwie dla każdego k .
Warunkowa heterozygotyczność
Obliczyliśmy warunkową heterozygotyczność dla każdej starożytnej populacji lewantyńskiej za pomocą popstats 75 . Do tej analizy wykorzystaliśmy zestaw danych HO +, ograniczając się do miejsc SNP ustalonych od pojedynczego osobnika Yoruba i do SNP transwersji, jak opisano w Skoglund i in. 44 .
f -statystyki
Obliczyliśmy statystyki f 4 za pomocą programu qpDstat w ADMIXTOOLS 35 z parametrami domyślnymi i trybem f 4 : TAK. Obliczyliśmy statystyki f 3 za pomocą programu qp3Pop w ADMIXTOOLS 35 , używając parametrów domyślnych, z chowem wsobnym: TAK. Przeprowadziliśmy wszystkie analizy przy użyciu zestawu danych HOIll +, z wyjątkiem statystyki f 4 (Levant_BA_North, Levant_BA_South; A, Chimp), którą przeprowadziliśmy na zbiorze danych HO +.
qpAdm
Oszacowaliśmy proporcje przodków w populacji Levant_ChL za pomocą metodologii qpAdm, z parametrami allsnps: TAK i szczegóły: TAK 36 . Przetestowaliśmy zarówno 2-, jak i 3-drożne domieszki między starożytnymi „lewicowymi” populacjami ze zbioru danych HOIll+. Wykorzystaliśmy populacje 09NW zdefiniowane w Lazaridis i in. 24 jako wstępne grupy zewnętrzne. Wybraliśmy dodatkowe grupy zewnętrzne na podstawie statystyk f 4 (Anatolia_N, Europa_EN; A, szympans) i f 4 (Levant_BA_North, Iran_ChL; A, szympans) i powtórzyliśmy qpAdm z każdą dodatkową grupą zewnętrzną dodaną do listy „Prawo”, aż wszystkie oprócz wyeliminowano jeden model z domieszką.
Użyliśmy qpAdm do określenia, czy populacje Levant_BA_South i Levant_BA_North można modelować przy użyciu Levant_ChL jako populacji źródłowej. Przetestowaliśmy dwukierunkowe domieszki między Levant_ChL i każdą inną starożytną „lewicową” populacją ze zbioru danych HOIll +. Przetestowaliśmy również „lewicowe” populacje Levant_N i Iran_ChL. Wykorzystaliśmy populacje 09NW „Right ” jako wstępne populacje grup zewnętrznych i potwierdziliśmy nasze ustalenia dla Levant_BA_North przy użyciu grup zewnętrznych zdefiniowanych w Haber et al. 26 . Dodaliśmy dodatkowe grupy zewnętrzne, aby jeszcze bardziej rozróżnić wiarygodne modele i powtarzaliśmy analizę qpAdm, aż wszystkie modele domieszek z wyjątkiem jednego zostały wyeliminowane.
qpWave
Obliczyliśmy minimalną liczbę strumieni przodków wymaganych do modelowania dwóch zestawów trzech populacji lewantyńskich (zestaw [1] Levant_N, Levant_ChL i Levant_BA_South, zestaw [2] Levant_N, Levant_BA_South, Levant_BA_North) przy użyciu metodologii qpWave 37, 38 z parametrem allsnps :TAK.
Porównania częstości alleli
Zbadaliśmy częstości SNP związane z fenotypowo ważnymi funkcjami w kategoriach metabolizmu, pigmentacji, podatności na choroby, odporności i stanu zapalnego w Levant_ChL w połączeniu z populacjami Levant_N, Levant_BA_North, Levant_BA_South, Anatolia_N i Iran_ChL, z częstościami alleli dla trzech połączonych populacji kontynentalnych populacje (AFR, EAS, EUR) w fazie 3 projektu 1000 genomów zgłoszone, jeśli są dostępne. Obliczyliśmy częstości alleli w każdym interesującym miejscu, obliczając prawdopodobieństwo częstości alleli referencyjnych populacji na podstawie danych, stosując metodę ustaloną w Mathieson i in. 31 . Dla każdej populacji o wielkości N obserwujemy sekwencje Ri , które posiadają allel referencyjny z całościT i sekwencje. Prawdopodobieństwo odniesienia częstości alleli p k , n , p ) = , w każdej populacji dane D = { X , N , R ja , T ja } wynosi L ( p ; D ) = gdzie B (∏ja = 1N{P2B (RI,TI, 1 – ε )+2p( 1 – p )b(RI,TI, 0.5 )+(1–str)2B (RI,TI, ε )}∏�=1�{�2�(��,��,1−ε)+2�(1−�)�(��,��,0,5)+(1−�)2�(��,��,ε)}(Nk)Pk( 1 − s)n – k(��)��(1−�)�−� wynosi dwumianowy rozkład prawdopodobieństwa, a to małe prawdopodobieństwo błędu, które dla naszych obliczeń ustawiliśmy na 0,001. Oszacowaliśmy częstości alleli, maksymalizując liczbowo prawdopodobieństwo dla każdej populacji.εε
Dostępność danych
Dopasowane sekwencje są dostępne w Europejskim Archiwum Nukleotydów pod numerem dostępu PRJEB27215. Zestawy danych genotypów wykorzystywane w analizie są dostępne pod adresem https://reich.hms.harvard.edu/datasets .
Zmiany w historii artykułu
05 września 2018 r
Ten artykuł został pierwotnie opublikowany bez załączonego pliku recenzji. Ten plik jest teraz dostępny w wersji HTML artykułu; PDF był poprawny od momentu publikacji.
20 września 2018 r
W oryginalnej wersji tego artykułu odniesienia w formacie „Pierwszy autor i in.”. zostały niewłaściwie usunięte. Błędy te zostały poprawione w wersji PDF i HTML artykułu
Bibliografia
-
Ussishkin, D. Świątynia Ghassulian w En-Gedi. Tel Awiw 7 , 1–44 (1980).
-
Seaton, P. Chalcolithic Cult and Risk Management w Teleilat Ghassul: The Area E Sanctuary (CMP (UK) Ltd., 2008).
-
Levy, TE Archeologia, antropologia i kult — sanktuarium w Gilat (Izrael, Londyn, 2006).
-
Perrot, J. & Ladiray, D. À Ossuaires De La Région Côtière Palestinienne Au Iv Millénaire Avant L’ère Chrétienne (Stowarzyszenie Paléorient, 1980).
-
van den Brink, WE w Shoham (North): Late Chalkolithic Burial Caves in the Lod Valley, Izrael (eds van den Brink, EC et al.) 175–190 (Israel Antiquities Authority, 2005).
-
Bar-Adon, P. Jaskinia skarbu e (The Israel Exploration Society, 1980).
-
Drabsch, B. Tajemnicze malowidła ścienne Teleilat Ghassul, Jordania (Archaeopress Archaeology, 2015).
-
Shalem, D. Ikonografia ossuariów i urn grobowych z późnego okresu chalkolitu w Izraelu w kontekście starożytnego Bliskiego Wschodu.Rozprawa doktorska, Haifa Univ. (2008).
-
Shalem, D. Motywy skarbu Nahal Mishmar i ossuariów: obserwacje porównawcze i interpretacje. J. Isr. Prehist. soc. 45 , 217–237 (2015).
-
Goren, Y. Gods & Caves, ORAZ uczeni Chalcolithic Cult and Metalurgy in the Judean Desert. Wschód. Archeol. (NEA) 77 , 260-266 (2014).
-
Tadmor, M. i in. Skarb Nahal Miszmar z Pustyni Judzkiej: technologia, skład i pochodzenie. Atiqot 27 , 95-148 (1995).
-
Anati, E. Palestyna przed Hebrajczykami (Alfred A. Knopf, 1963).
-
de Vaux, R. w Cambridge Historia starożytna, tom. 1 (red. Gadd, CJ, Edwards, IES i Hammond, NGL) 498–538 (Cambridge University Press, 1970).
-
Bourke, SJ w The Prehistory of Jordan II: Perspectives From 1997, Studies in Early near Eastern Production, Subsistence, and Environment (red. Kafafi, Z., Rollefson, G. & Gebel, HGK) 395–417 (Ex oriente, 1997) .
-
Bourke, SJ w badaniach z historii i archeologii Jordanii, tom. 6 (red. Zaghloul, I.) 249-259 (Departament Starożytności, 1997).
-
Gilead, I. Okres chalkolitu w Lewancie. J. Prehist świata. 2 , 397-443 (1988).
-
Hennessy, JB Wstępny raport z pierwszego sezonu wykopalisk w Teleilat Ghassul. Lewant 1 , 1–24 (1969).
-
Levy, TE w Archeologia społeczeństwa w Ziemi Świętej (red. Levy, TE) 226–244 (Leicester University Press, 1995).
-
Moore, AM Późny neolit w Palestynie. Lewant 5 , 36-68 (1973).
-
Gal, Z., Smithline, H. & Shalem, D. Chalkolityczna jaskinia grobowa w Peqi’in, Górna Galilea. Isr. Eksploruj. J. 47 , 145-154 (1997).
-
Shalem, D. i in. Peqi’in: A Late Chalcolithic Burial Site Upper Galilee, Izrael (Kinneret Academic College Institute for Galilean Archaeology, 2013).
-
Nagar, Y. w Peqi ’ w : A. Late Chalkolithic Burial Site, Upper Galilee, Izrael (red. Shalem. D., Gal, Z. & Smithline, H.) 391–405 (The Institute for Galilean Archaeology, 2013).
-
Segal, D., Carmi, I., Gal, Z., Smithline, H. & Shalem, D. Datowanie chalkolitycznej jaskini grobowej w Peqi’in, górna Galilea, Izrael. Radiowęgiel 40 , 707-712 (1998).
-
Lazaridis, I. i in. Wgląd genomowy w pochodzenie rolnictwa na starożytnym Bliskim Wschodzie. Przyroda 536 , 419 (2016).
-
Broushaki, F. i in. Genomy wczesnego neolitu ze wschodniego Żyznego Półksiężyca. Nauka 353 , 499-503 (2016).
-
Haber, M. i in. Ciągłość i domieszka w ciągu ostatnich pięciu tysiącleci historii Lewantynu od starożytnych kananejskich i współczesnych sekwencji genomu libańskiego. Jestem. J. Hum. Genet. 101 , 274-282 (2017).
-
Gamba, C. i in. Przepływ genomu i zastój w pięciotysięcznym transekcie europejskiej prehistorii. Nat. Komuna. 5 , 5257 (2014).
-
Dabney, J. i in. Kompletna sekwencja genomu mitochondrialnego niedźwiedzia jaskiniowego ze środkowego plejstocenu zrekonstruowana z ultrakrótkich fragmentów DNA. proc. Natl Acad. nauka 110 , 15758-15763 (2013).
-
Rohland, N., Harney, E., Mallick, S., Nordenfelt, S. & Reich, D. Częściowe leczenie uracylowo-DNA-glikozylazą do badań przesiewowych starożytnego DNA. Filoz. Trans. R. Soc. B370 , 20130624 (2015).
-
Fu, Q. i in. Analiza DNA wczesnego współczesnego człowieka z jaskini Tianyuan w Chinach. proc. Natl Acad. nauka 110 , 2223-2227 (2013).
-
Mathieson, I. i in. Wzorce selekcji obejmujące cały genom u 230 starożytnych Eurazjatów. Natura 528 , 499–503 (2015).
-
Korneliussen, TS, Albrechtsen, A. & Nielsen, R. ANGSD: analiza danych sekwencjonowania nowej generacji. BMC Bioinformatyka. 15 , 356 (2014).
-
Patterson, N., Price, AL & Reich, D. Struktura populacji i analiza własna. PLoS. Genet. 2 , e190 (2006).
-
Alexander, DH, Novembre, J. & Lange, K. Szybka oparta na modelu ocena pochodzenia u niepowiązanych osób. Genom Res. 19 , 1655-1664 (2009).
-
Patterson, N. i in. Starożytna domieszka w historii ludzkości.Genetyka 192 , 1065-1093 (2012).
-
Haak, W. i in. Masowa migracja ze stepu była źródłem języków indoeuropejskich w Europie. Natura 522 , 207–211 (2015).
-
Moorjani, P. i in. Genetyczne dowody na niedawną mieszankę populacji w Indiach. Jestem. J. Człowiek. Genet. 93 , 422-438 (2013).
-
Reich, D. i in. Rekonstrukcja historii populacji rdzennych Amerykanów. Przyroda 488 , 370–374 (2012).
-
Konsorcjum Projektu 1000 Genomów. Globalne odniesienie do zmienności genetycznej człowieka. Przyroda 526 , 68 (2015).
-
Eiberg, H. i in. Niebieski kolor oczu u ludzi może być spowodowany doskonale powiązaną mutacją założycielską w elemencie regulatorowym zlokalizowanym w genie HERC2 hamującym ekspresję OCA2. Szum. Genet. 123 , 177-187 (2008).
-
Soejima, M. & Koda, Y. Różnice w populacji dwóch kodujących SNP w genach związanych z pigmentacją SLC24A5 i SLC45A2. Int. J. Noga. Med. 121 , 36-39 (2007).
-
Martin, AR i in. Nieoczekiwanie złożona architektura pigmentacji skóry u Afrykanów. Cela 171 , 1340–1353 (2017).
-
Rowan, YM & Golden, J. Okres chalkolitu południowego Lewantu: przegląd syntetyczny. J. Prehistoria świata 22 , 1–92 (2009).
-
Skoglund, P. i in. Wgląd genomowy w zaludnienie południowo-zachodniego Pacyfiku. Przyroda 538 , 510 (2016).
-
Skoglund, P. i in. Rekonstrukcja prehistorycznej struktury populacji Afryki. Cela 171 , 59–71 (2017).
-
Mendez, FL i in. Zwiększona rozdzielczość haplogrupy T chromosomu Y określa relacje między populacjami Bliskiego Wschodu, Europy i Afryki. Szum. Biol. 83 , 39-53 (2011).
-
Shalem, D. Ciągłość kulturowa i zmiany w późnych chalkolitycznych zwyczajach pogrzebowych i obrazach ikonograficznych w południowym Lewantynie: interpretacja znalezisk z jaskini Peqi’in. J. Isr. Prehist. soc. 47 , 148-170 (2017).
-
Merhav, R., Heltzer, M., Segal, A. & Kaufman, D. w badaniach z archeologii i historii starożytnego Izraela na cześć Mosze Dothana (red. Heltzer, M., Segal, A. & Kaufman, D. .) 21–42 (Haifa University Press, 1993).
-
Bar-Yosef, O. & Ayalon, E. Chalkolityczne ossuaria: co imitują i dlaczego? Kadmoniot 34 , 34-43 (2001).
-
Beck, P. w Essays in Ancient Civilization przedstawionym Helene J. Kantor. Studies in Ancient Oriental Civilization (red. Leonard, A. & Williams, BB) 39–54 (The Oriental Institute, 1989).
-
Yahalom-Mack, N. i in. Najwcześniejszy ołów obiekt w Levant. PLoS JEDEN 10 , e0142948 (2015).
-
Gilead, I. Historia osadnictwa chalkolitycznego w rejonie Nahal Beer Sheva: aspekt radiowęglowy. Byk. Jestem. Sch. Orient. Rez. 296 , 1-13 (1994).
-
Bourke, S. i in. Chronologia okresu Ghassulian Chalcolithic w południowym Lewancie: nowe oznaczenia 14 C z Teleilat Ghassul w Jordanii. Radiowęgiel 43 , 1217-1222 (2001).
-
Bourke, SJ & Lovell, JL Ghassul, chronologia i sekwencjonowanie kulturowe. Paléorient 30 , 179-182 (2004).
-
Gilead, I. in Culture, Chronology and the Chalcolith: Theory and Transition (red. Lovell, JL & Rowan, YM) 12–24 (Oxbow Books, 2011).
-
van den Brink Edwin, C. in Culture, Chronology and the Chalcolith: Theory and Transition (red. Lovell, JL & Rowan, YM) 61–70 (Oxbow Books, 2011).
-
Vardi, J. & Gilead, I. Chalkolityczna wczesna epoka brązu I przejście w południowym Lewancie: perspektywa litu. Paléorient 39 , 111–123 (2013).
-
Milevski, I. Przejście od epoki chalkolitu do wczesnej epoki brązu w południowym Lewancie w kontekście społeczno-ekonomicznym. Paléorient 39 , 193–208 (2013).
-
Braun, E. & Roux, V. Przejście z późnego chalkolitu do wczesnej epoki brązu I w południowym Lewancie: określenie ciągłości i nieciągłości lub „Uważaj na lukę”. Paléorient 29 , 15–22 (2013).
-
Joffe, AH & Dessel, J. Redefinicja chronologii i terminologii dla chalkolitu południowego Lewantu. bież. Antropol. 36 , 507-518 (1995).
-
Yadin, Y. Najwcześniejsza wzmianka o militarnej penetracji Egiptu do Azji? Niektóre aspekty palety Narmera, „pustynnych latawców” i mezopotamskich cylindrów z pieczęciami. Isr. Eksploruj. J. 5 , 1-16 (1955).
-
Yeivin, S. Wczesne kontakty między Kanaanem a Egiptem. Isr. Eksploruj. J. 10 , 193-203 (1960).
-
Ussishkin, D. „Ghassulska” świątynia w Ein Gedi i geneza skarbu z Nahal Miszmar. Archeol biblijny. 34 , 23-39 (1971).
-
Davidovich, U. Chalkolityczna wczesna epoka brązu: widok z jaskiń Pustyni Judzkiej, Lewant Południowy. Paléorient , 39 , 125–138 (2013).
-
Korlević, P. i in. Zmniejszenie zanieczyszczenia mikrobiologicznego i ludzkiego w ekstrakcjach DNA ze starożytnych kości i zębów. Biotechniki 59 , 87–93 (2015).
-
Fu, Q. i in. Wczesny współczesny człowiek z Rumunii z niedawnym przodkiem neandertalczykiem. Przyroda 524 , 216–219 (2015).
-
Harbison, CT i in. Transkrypcyjny kod regulacyjny genomu eukariotycznego. Przyroda 431 , 99-104 (2004).
-
Li, H. & Durbin, R. Szybkie i dokładne wyrównanie krótkiego odczytu z transformacją Burrowsa-Wheelera. Bioinformatyka 25 , 1754–1760 (2009).
-
Behar, DM i in. „Kopernikańska” ponowna ocena ludzkiego drzewa mitochondrialnego DNA od jego korzenia. Jestem. J. Człowiek. Genet. 90 , 675–684 (2012).
-
Weissensteiner, H. i in. HaploGrep2: mitochondrialna klasyfikacja haplogrup w dobie wysokowydajnego sekwencjonowania. Kwasy nukleinowe Res. 44 , W58–W63 (2016).
-
Skoglund, P. i in. Oddzielanie starożytnego DNA od współczesnego skażenia u syberyjskiego neandertalczyka. proc. Natl Acad. nauka 111 , 2229-2234 (2014).
-
Poznik, GD i in. Przerywane wybuchy w męskiej demografii człowieka wywnioskowano z 1244 światowych sekwencji chromosomu Y. Nat. Genet. 48 , 593 (2016).
-
Kuhn, JMM, Jakobsson, M. & Günther, T. Szacowanie genetycznych pokrewieństwa w populacjach prehistorycznych. PLoS JEDEN 13 , e0195491 (2018).
-
Purcell, S. i in. PLINK: zestaw narzędzi do asocjacji całego genomu i analiz powiązań opartych na populacji. Jestem. J. Człowiek. Genet. 81 , 559-575 (2007).
-
Skoglund, P. i in. Dowody genetyczne dla dwóch założycielskich populacji obu Ameryk. Przyroda 525 , 104 (2015).
Podziękowanie
Jaskinia grobowa Peqi’in została wykopana pod auspicjami Izraelskiego Urzędu Starożytności. EH był wspierany przez stypendium dla absolwentów z Max Planck-Harvard Research Center for the Archaeoscience of the Ancient Mediterranean (MHAAM). DR był wspierany przez grant HOMINID amerykańskiej Narodowej Fundacji Nauki BCS-1032255, grant US National Institutes of Health GM100233, grant Allen Discovery Center i jest badaczem Instytutu Medycznego Howarda Hughesa. Badanie antropologiczne było wspierane przez Fundację Dana Davida. Dziękujemy Vagheeshowi Narasimhanowi za wygenerowanie i opisanie wywołań haplogrupy chromosomu Y. Dziękujemy Arielowi Pokhojaevowi za stworzenie obrazu mapy użytego na ryc. 1a . Dziękujemy Johnowi Wakeleyowi za krytyczne uwagi.
Deklaracje etyczne
Konkurujące interesy
Autorzy deklarują brak sprzecznych interesów.
Dodatkowe informacje
Uwaga wydawcy: Springer Nature pozostaje neutralny w odniesieniu do roszczeń jurysdykcyjnych na opublikowanych mapach i powiązań instytucjonalnych.
Harney, É., May, H., Shalem, D. i in. Starożytne DNA z chalkolitycznego Izraela ujawnia rolę mieszanki populacji w transformacji kulturowej. Nat Commun 9 , 3336 (2018). https://doi.org/10.1038/s41467-018-05649-9.
Link do artykułu: https://www.nature.com/articles/s41467-018-05649-9#article-info
Prawa i uprawnienia
Otwarty dostęp Ten artykuł jest objęty licencją Creative Commons Attribution 4.0 International License, która zezwala na używanie, dzielenie się, adaptację, dystrybucję i powielanie na dowolnym nośniku lub w dowolnym formacie, pod warunkiem, że podasz odpowiednie oznaczenie oryginalnego autora (autorów) i źródła, podać link do licencji Creative Commons i wskazać, czy dokonano zmian. Obrazy lub inne materiały stron trzecich zawarte w tym artykule są objęte licencją Creative Commons, chyba że zaznaczono inaczej w informacji o autorze materiału. Jeśli materiał nie jest objęty licencją Creative Commons, a zamierzone użycie jest niezgodne z przepisami prawa lub wykracza poza dozwolone użycie, musisz uzyskać pozwolenie bezpośrednio od właściciela praw autorskich. Aby zobaczyć kopię tej licencji, odwiedźhttp://creativecommons.org/licenses/by/4.0/.
Obraz wyróżniający: Chalkolityczna kopalnia miedzi w dolinie Timna, pustynia Negew, Izrael. Z Wikimedia Commons, repozytorium wolnych multimediów